refine.bio, Part 2
TL;DR: We need to put together lots of data from disparate sources at low cost via cloud workflows, and this technical post describes the CCDL’s system using specific examples. The refinery runs on AWS and uses Docker and Nomad. Its source code is available on GitHub.
Table of Contents
- Introduction
- The Surveyor System
- The Message System
- The Downloaders System
- The Processors System
- Conclusion
1. Introduction
In refine.bio, Part 1 I explained the need for refine.bio along with many of the specific requirements it would need to meet. In this post I will explain the design I came up to meet those requirements, implementation details about it, and then examples that follow how data is surveyed, downloaded, and then processed.
The design of refine.bio is focused around two abstractions: batches and jobs. A batch is a singular unit of data to be processed. It could be a sample, a genome, or another entity. A job is a unit of work that the system does. There are three kinds of jobs:
- Survey Job: A job that discovers new batches.
- Downloader Job: A job that downloads the data for one or more batches.
- Processor Job: A job that modifies data associated with one or more batches.
The most natural way to discuss the design of refine.bio is in terms of these two abstractions because they represent the core functionality of the system: it refines (via jobs) data (represented by batches). Making these concepts reified entities enables the system to record what it has done and when. The batches table records what data is available, where it can be downloaded from, and any other information the system has about the data. The tables for the various jobs will record when the data was discovered, when it was downloaded, and what processing steps the system performed on it. This will enable a UI to be built exposing this information to users, which will provide them with the context necessary to trust the fidelity of the data.
To help explain how this all comes together I will walk through an example I recently implemented which built Salmon indices for every organism contained in Ensembl Genomes. Salmon is a tool for analyzing RNASeq data which we are using in refine.bio to quantify transcripts. In order to be able to use Salmon, you first need a Salmon index for your transcriptome. Each Salmon index is specific for one organism, so we decided to use refine.bio to preprocess all the indices ahead of time. The basic outline of what we did for each organism is:
- Download the organism’s genome from Ensembl.
- Clean up the genome annotation file.
- Use RSEM to build a custom transcriptome.
- Feed that custom transcriptome into the salmon index command.
We’ll use this diagram as a map to follow our example as the data goes from being stored in Ensembl to being a fully processed Salmon index. I will note here that the code examples used in this post have been stripped down as much as possible to make them easy to read and understand. Therefore almost all of the housekeeping and error handling parts of the code have been omitted along with many comments.
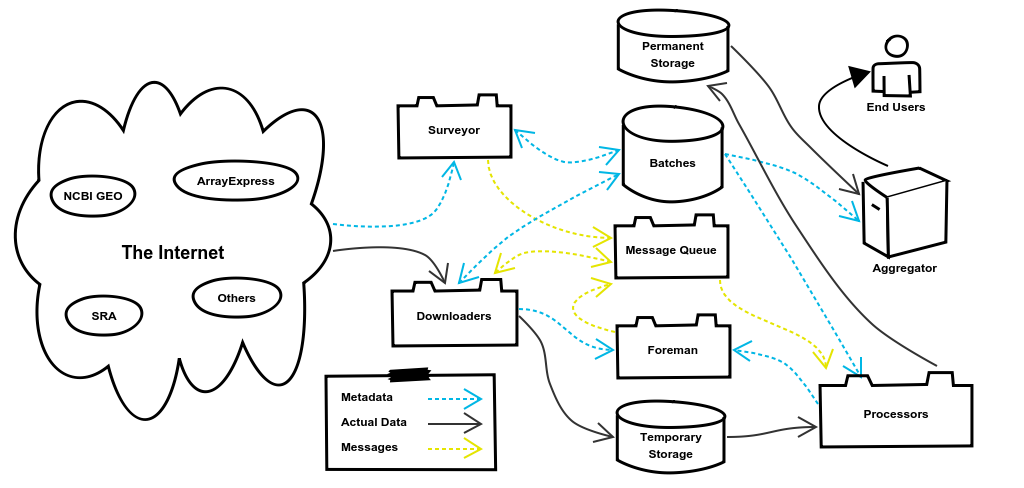
2. The Surveyor System
We’re going to start by looking at how the Surveyor pulls metadata from Ensembl, creates Batches, and then queues DownloaderJobs. The way to create a new Surveyor is to create an implementation of the abstract ExternalSourceSurveyor class. ExternalSourceSurveyorhas three methods which we will need to override:
- discover_batches() - Discovers Batches and add them to the surveyor’s list of Batches by calling theExternalSourceSurveyor.add_batch() method.
- determine_pipeline() - Specifies which Processor Pipeline should run on each Batch. This will be stored as the pipeline_required property on the batches table.
- group_batches() - Groups Batches together that should be downloaded together.
The way these methods will be used together is best demonstrated by looking at the ExternalSourceSurveyor.survey() method:
def survey(self):
self.discover_batches()
for group in self.group_batches():
self.queue_downloader_jobs(group)
So what happens there is that ExternalSourceSurveyor and classes which inherit from it have a property self.batches which is a list of Batches. The discover_batches() method is expected to populate that property via the add_batch() method. For each Batchdiscover_batches() discovers, it calls add_batch(). This function takes required fields for the Batch table, a list of Files associated with the Batch, and a dictionary of key-value pairs which are tacked onto the Batch.
So let’s take a look at what our example’s discover_batches() method looks like:
def discover_batches(self):
ensembl_division = (
SurveyJobKeyValue
.objects
.get(survey_job_id=self.survey_job.id,
key__exact="ensembl_division")
.value
)
r = requests.get(DIVISION_URL_TEMPLATE.format(division=ensembl_division))
# Yes I'm aware that specieses isn't a word. However, I need to
# distinguish between a singular species and multiple species.
specieses = r.json()
for species in specieses:
self._generate_batches(species)
To break down what’s going on: the division of Ensembl which we’re surveying in this particular job is retrieved, a list of species is pulled from Ensembl’s REST API and a Batch is generated for each species. There are quite a few idiosyncrasies when working with the collection of APIs and FTP servers which make up Ensembl Genomes, so I’m not going to delve into everything _generate_batches() does. However, it primarily builds the appropriate URL to download the Batch’s files, creates File objects for both (GTF and FASTA) file types, and cleans up some of the metadata returned by Ensembl’s REST API. However, the most important thing it does is call ExternalSourceSurveyor.add_batch() like so:
self.add_batch(platform_accession_code=platform_accession_code,
experiment_accession_code=url_builder.file_name_species.upper(),
organism_id=url_builder.taxonomy_id,
organism_name=url_builder.scientific_name,
experiment_title="NA",
release_date=current_time,
last_uploaded_date=current_time,
files=[fasta_file, gtf_file],
key_values=species)
add_batch() then takes care of preventing duplicate Batches, creating the Batch objects, setting the correct Processor Pipeline (by calling the user-defined determine_pipeline() method), and saving everything to the database. For this source we only run one Processor Pipeline so we simply return the correct enumerated value. Therefore the implementation of determine_pipeline() is a single line that looks like this:
def determine_pipeline(self, batch: Batch, key_values: Dict = {}) -> ProcessorPipeline:
return ProcessorPipeline.TRANSCRIPTOME_INDEX
At this point we’ve implemented our determine_pipeline() and discover_batches() methods, leaving only group_batches(). Now there’s one detail about the Salmon Indices we’re building that I haven’t yet mentioned: we need two for each organism. We need one that is built to handle samples with a long average transcript read length and one for a short average transcript read length. The way we do this is to have _generate_batches() actually generate two Batches which are identical except that we add a ”kmer_size” key to the key_values dictionary that we pass through to add_batch(). However, both Batches need the same files to run. This setup allows us to only download them once.
We’re able to do this by creating a single DownloaderJob for both Batches. This is exactly what the ExternalSourceSurveyor.group_batches() method is for. We can group the Batches based on the download URL of their Files like so:
def group_batches(self) -> List[List[Batch]]:
"""Groups batches based on the download URL of their first File."""
download_url_mapping = {}
for batch in self.batches:
download_url = batch.files[0].download_url
if download_url in download_url_mapping:
download_url_mapping[download_url].append(batch)
else:
download_url_mapping[download_url] = [batch]
return list(download_url_mapping.values())
If we look back at the ExternalSourceSurveyor.survey() method:
def survey(self):
self.discover_batches()
for group in self.group_batches():
self.queue_downloader_jobs(group)
It will now queue DownloaderJobs for each of the groups returned by the group_batches() method we defined. At this point the Surveyor we’ve implemented will:
- Discover what species are stored in Ensembl.
- Record metadata and create two Batches for each species.
- Queue DownloaderJobs for each group of Batches.
This covers the responsibilities of the Surveyor system. In just a little bit we’ll take a look at what the the Downloaders System does with the jobs we’ve sent it. But first we’ll take a look at how the different systems send messages to each other.
3. The Message System
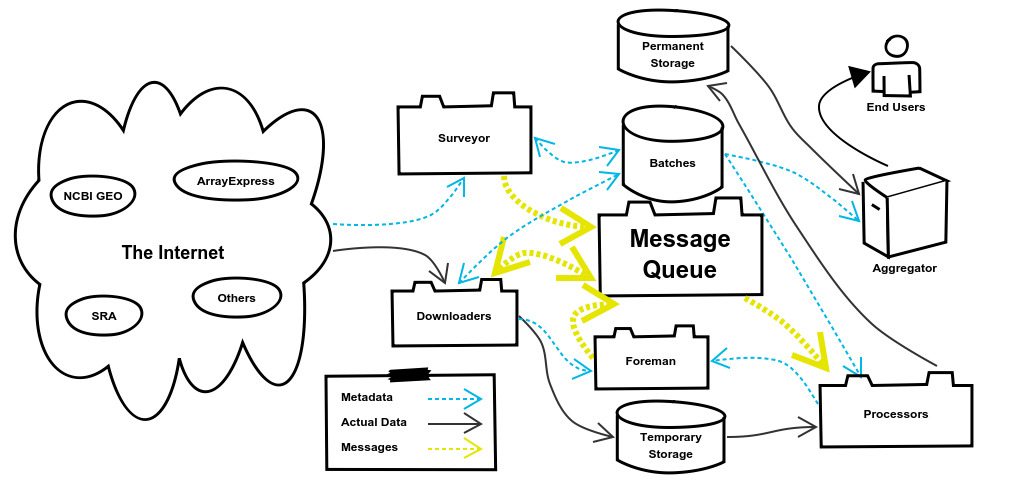
The Message System of refine.bio allows different components to communicate with each other. In the last section I mentioned that the Surveyor System queues DownloaderJobs, but I didn’t define what that meant. Queuing a DownloaderJob has two parts to it. The first creates a database entry representing the DownloaderJob. This entry records information about the job such as when it started, when it ended, and if it was successful. This allows the Foreman to check up on jobs to see if they need to be requeued and is preserved in case it is ever needed to verify the fidelity of a particular Batch of data.
The second part of queuing a DownloaderJob is to send a message to the Downloaders System so it knows that it has work to do. This is how both of these things happen:
@retry(stop_max_attempt_number=3)
def queue_downloader_jobs(self, batches: List[Batch]):
downloader_job = DownloaderJob.create_job_and_relationships(
batches=batches, downloader_task=self.downloader_task())
try:
send_job(downloader_task, downloader_job.id)
except:
# If the task doesn't get sent we don't want the
# downloader_job to be left floating
downloader_job.delete()
raise
The DownloaderJob.create_job_and_relationships() static method creates and saves a DownloaderJob to the database, along with creating links to each of the Batches it should process. Once that is done we send a message to the Downloaders System through the Message System via send_job(). If a failure occurs for any reason, such as a network partition, we delete the DownloaderJob so it’s not left in the database without a message telling the Downloaders System to pick it up. This entire method is wrapped with the decorator @retry(stop_max_attempt_number=3) to attempt to overcome temporary failures.
So, we send a message through the Message System to tell the Downloaders System to pick up the job, but how does that actually work? The Message System is not actually something I’ve built because distributed message queues are a hard problem that a lot of other people have expended a lot of effort to get right. There are a number of tools that could be used for the job such as RabbitMQ, Amazon SQS, or Apache Kafka. We use Hashicorp’s Nomad to manage messages.
Our decision to switch to Nomad was actually unrelated to the Message System. Nomad isn’t actually a message queue but it can be used as one for our purposes. We needed an orchestration tool to manage the cluster we’re going to use for the Downloaders System and the Processors System. Nomad works by running an agent on each node of the cluster. This agent does two primary things: it communicates with the rest of the cluster and it performs fingerprinting on the node. This fingerprinting discovers how many resources the node has available to it such as RAM, disk space, and CPU power. This allows the leader nodes to receive a job message and find a node with the available resources needed to run that job, whether it’s a DownloaderJob or a ProcessorJob. By carefully estimating the amount of resources each job will require, we efficiently use the cluster’s computing power. Nomad’s scheduling system then aims to solve a form of the bin packing problem where jobs are objects with different volumes and the nodes are the bins we want to pack as tightly as possible.
So when I discuss sending a message to the Message System what actually happens is that a Nomad job message is sent to the Nomad leaders, which then find an appropriate node of the cluster to perform the work. However, most contributors to the project won’t have to think too much about what is happening under the hood; they can just send a message to the Message System and trust it to find a node to perform the work associated with that job. So now that we know how the message that our Surveyor sends manages to make it to the Downloaders System we can dive into what that system does.
4. The Downloaders System
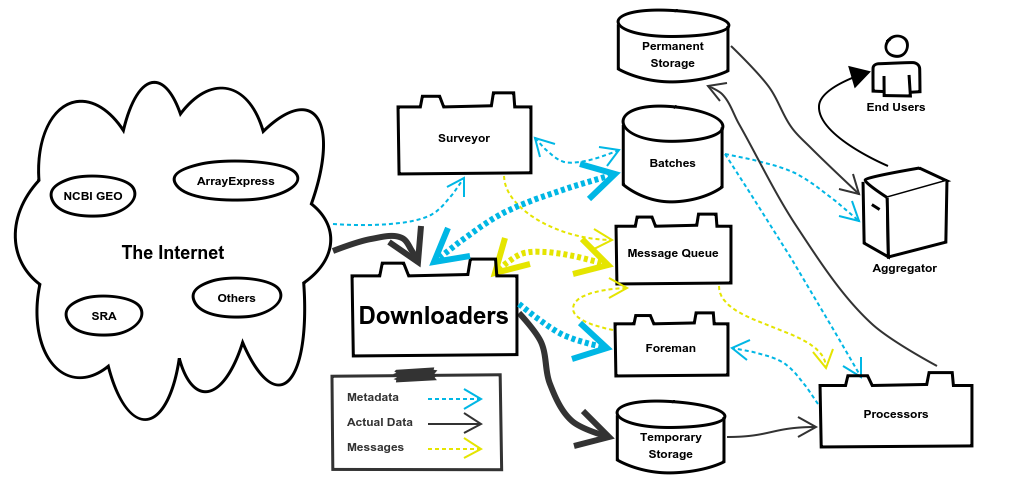
The Downloaders System is responsible for downloading the data for Batches from external sources. Once the data is downloaded it will be stored in the Temporary Storage, which we’re using AWS S3 for. When it comes time to process the data the Processors System, pulls it back out of Temporary Storage. It may seem inefficient to download the data, push it to Temporary Storage, and then pull it back out to process it. Why not just download it and then immediately process it without that other step? There are a couple reasons for this.
If the ProcessorJob fails for any reason we won’t have to re-download the data. Once a DownloaderJob has completed successfully we won’t need to re-download the data from the external source, no matter what issues arise while processing it. Also, this lets us use resources more efficiently. This ties into what I discussed in the last section about how Nomad’s fingerprinting and scheduling capabilities can be used to achieve efficient bin packing. DownloaderJobs have very low resource requirements but can take a while because we can only download data as fast as external servers will provide the data. ProcessorJobs on the other hand tend to have much higher resource requirements because of the processing work we’re performing. If we did not separate these two responsibilities then our MegaJobswould need to be allocated a large number of resources, which they would then not utilize for a long time while downloading the data from external sources. By having ProcessorJobs download the data from within the system, we get the benefit of the good network latency between AWS EC2 and S3.
With your burning questions of “why split those responsibilities?!?!?” answered, we can move on to what a Downloader for our example looks like. I am going to make use of the functions _download_file(download_url: str, file_path: str, job: DownloaderJob) and _upload_files(job_dir: str, files: List[File], job: DownloaderJob) without showing the code for them. This is because they pretty much just do what their names imply; most of their actual implementation involves handling and logging network errors. So here’s the main function for the Downloader:
def download_transcriptome(job_id: int) -> None:
# Block One
job = utils.start_job(job_id)
batches = job.batches.all()
# Prevents different jobs from using the same directory
job_dir = "downloader_job_" + str(job_id)
# Block Two
first_fasta_file = File.objects.get(batch=batches[0], raw_format__exact="fa.gz")
first_gtf_file = File.objects.get(batch=batches[0], raw_format__exact="gtf.gz")
second_fasta_file = File.objects.get(batch=batches[1], raw_format__exact="fa.gz")
second_gtf_file = File.objects.get(batch=batches[1], raw_format__exact="gtf.gz")
os.makedirs(first_fasta_file.get_temp_dir(job_dir), exist_ok=True)
# The two Batches share the same fasta and gtf files, so
# only download each one once
_download_file(first_fasta_file.download_url,
first_fasta_file.get_temp_pre_path(job_dir),
job)
_download_file(first_gtf_file.download_url,
first_gtf_file.get_temp_pre_path(job_dir),
job)
# Block Three
# Then create symlinks so the files for the second Batch
# can be found where they will be expected to.
os.symlink(first_fasta_file.get_temp_pre_path(job_dir),
second_fasta_file.get_temp_pre_path(job_dir))
os.symlink(first_gtf_file.get_temp_pre_path(job_dir),
second_gtf_file.get_temp_pre_path(job_dir))
_upload_files(job_dir,
[first_fasta_file, first_gtf_file, second_fasta_file, second_gtf_file],
job)
utils.end_job(job, batches, success=True)
This function is pretty long and there’re a few different parts so I’ve stuck in a few comments denoting different code blocks. Block One is just getting the job started. It calls utils.start_job() which will mark the job as started and retrieve the DownloaderJob object from the database. We then retrieve the Batches from the database and build our job_dir_prefix which we will put in every path we use during this job so we don’t overlap with any other jobs running concurrently on the same instance. In Block Two we retrieve the File objects out of the database, create a temporary directory to download files into, and then we actually download one set of files.
In Block Three we deal with the fact that we need the same files for two different batches. This goes back to what I discussed in the Surveyor System section about building two Salmon Indices for each organism. Rather than just re-download the exact same files again, we create symlinks to the locations that those files are expected to have been downloaded to. That’s what the File.get_temp_pre_path() method does, it returns where the path where the unprocessed file should be located (pre as in pre-processing). Now when we call the _upload_files() function it will have each of the Files we passed into it upload themselves. That’s right, each File object is capable of uploading itself to Temporary Storage via its upload_raw_file() method. Finally we just call utils.end_job() to mark the job as completed successfully. (In the actual function we catch errors and call utils.end_job() with success=False when appropriate.)
The one other thing that utils.end_job() will take care of for us is queuing a ProcessorJob. This is very similar to the way a DownloaderJob was queued earlier, so I’ll just move along to…
5. The Processors System
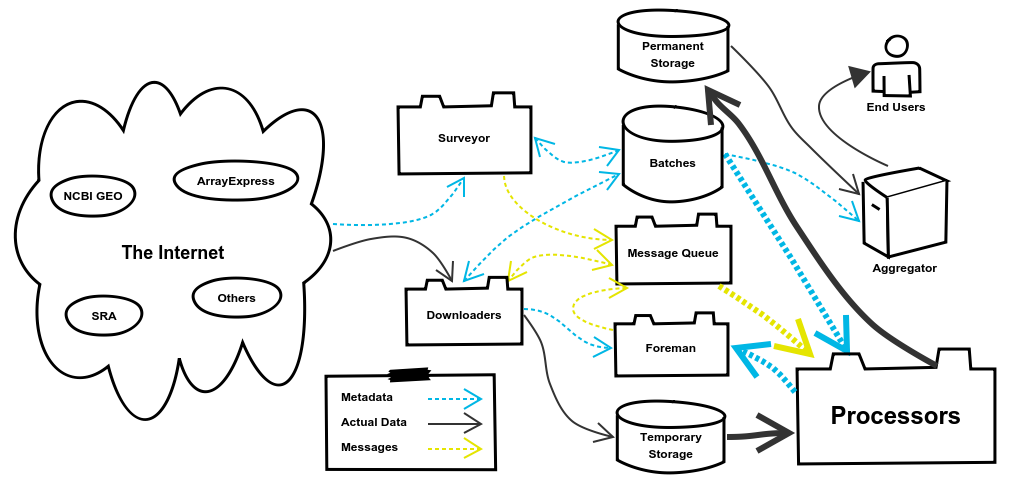
You’ve made it to the good stuff! The Processors System puts the refine in refine.bio. The full range of types of processing that we actually perform is outside the scope of this blog post, but they are carefully considered by the project’s Scientific Lead, Dr. Jaclyn Taroni. Instead we’ll focus on just the example we’ve been following throughout this post. The basic flow for all Processors is to retrieve the Batch’s data from Temporary Storage, perform some kind of processing, and then push the processed data into Permanent Storage. The processing we’ll need to perform for our example are the steps I outlined in the beginning of the post:
- Clean the genome annotation file.
- Use RSEM to build an custom transcriptome.
- Feed that custom transcriptome into the salmon index command.
However, before we can dive into actually doing all of that we need to discuss what Processors are. A Processor is actually just a function that accepts a single argument: the ID of a ProcessorJob. When the Processor System receives a job message from the Message System it will extract the ProcessorJob ID from the message along with the name of the Processor to execute. It then runs the function and waits for it to complete so it can report that the job’s computational resources can be used by other jobs.
Despite the minimal specification of Processors, there are certain responsibilities they are expected to complete. Each Processor must mark when it starts the job, when it ends the job, and whether or not the job was successful. They should also free up any disk space they utilize and if they’re successful they should remove the raw data from Temporary Storage. Since every Processor needs to do this stuff a framework is provided by the system to make it easy and to keep things DRY. The framework was designed with the goal of allowing reusable and composable functions to be created which can then be joined together into a single pipeline. Each function in the pipeline takes a job_context dictionary as an input and must output another (or the same) dictionary which can be passed into the next function of the pipeline. This behavior can be utilized by calling processors.utils.run_pipeline(), which takes two arguments: an initial job_context dictionary and a list of functions.
To illustrate this let’s take a look at the Processor function for our example:
def build_index(job_id: int) -> None:
utils.run_pipeline({"job_id": job_id},
[utils.start_job,
_set_job_prefix,
_prepare_files,
_process_gtf,
_create_index,
_zip_index,
utils.upload_processed_files,
utils.cleanup_raw_files,
utils.end_job])
Any function which returns a job_context containing ”success”: False will cause the pipeline to terminate and the job to be marked as a failure. Notice that the list of functions is composed of calls to functions defined in the utils namespace and private functions from the current namespace. The functions defined in the utils namespace take care of the common functionality I discussed in the previous paragraph. They pretty much all do what their names imply, but it will be helpful to note that in addition to marking the ProcessorJob as started, utils.start_job() also retrieves the ProcessorJob and Batch objects from the database and adds them to the job_context. So now let’s take a look at our pipeline step by step starting with:
def _set_job_prefix(job_context: Dict) -> Dict:
job_context["job_dir_prefix"] = "processor_job_" + str(job_context["job_id"])
return job_context
This adds the key “job_dir_prefix” to the job_context, which serves a similar purpose to the variable named similarly from the Downloaders System section: it makes all paths related to this job unique on the local disk. Next we run _prepare_files() which has the purpose of pulling the files from Temporary Storage and unzipping them so they’re ready to be processed.
def _prepare_files(job_context: Dict) -> Dict:
# Transcriptome index processor jobs have only one batch. Each
# batch has a fasta file and a gtf file
batch = job_context["batches"][0]
fasta_file = File.objects.get(batch=batch, raw_format__exact="fa.gz")
gtf_file = File.objects.get(batch=batch, raw_format__exact="gtf.gz")
fasta_file.download_raw_file(job_context["job_dir_prefix"])
gtf_file.download_raw_file(job_context["job_dir_prefix"])
gzipped_fasta_file_path = fasta_file.get_temp_pre_path(job_context["job_dir_prefix"])
gzipped_gtf_file_path = gtf_file.get_temp_pre_path(job_context["job_dir_prefix"])
job_context["fasta_file_path"] = gzipped_fasta_file_path.replace(".gz", "")
job_context["gtf_file_path"] = gzipped_gtf_file_path.replace(".gz", "")
with gzip.open(gzipped_fasta_file_path, "rb") as gzipped_file, \
open(job_context["fasta_file_path"], "wb") as gunzipped_file:
shutil.copyfileobj(gzipped_file, gunzipped_file)
with gzip.open(gzipped_gtf_file_path, "rb") as gzipped_file, \
open(job_context["gtf_file_path"], "wb") as gunzipped_file:
shutil.copyfileobj(gzipped_file, gunzipped_file)
job_context["fasta_file"] = fasta_file
job_context["gtf_file"] = gtf_file
return job_context
With our files unzipped locally, we’re ready to start processing by calling process_gtf(). This function cleans the genome annotation file (the gtf_file in this case) and creates a mapping from genes to transcripts for the given genome. If you’re not sure what that means, don’t worry about it; the upshot is that job_context["gtf_file_path"] is updated to point to the cleaned GTF file and the ”genes_to_transcripts_path” key is added to the job_context. Now we’re ready to create a Salmon Index using RSEM and Salmon:
def _create_index(job_context: Dict) -> Dict:
# Prepare the output directories.
work_dir = job_context["gtf_file"].get_temp_dir(job_context["job_dir_prefix"])
job_context["output_dir"] = os.path.join(work_dir, "index")
rsem_index_dir = os.path.join(work_dir, "rsem_index")
os.makedirs(rsem_index_dir, exist_ok=True)
# RSEM takes a prefix path and then all files generated by it will
# start with that
rsem_prefix = os.path.join(rsem_index_dir,
job_context["fasta_file"].get_base_name())
rsem_command_string = (
"rsem-prepare-reference --gtf {gtf_file}"
" --transcript-to-gene-map {genes_to_transcripts} {fasta_file} {rsem_prefix}"
)
rsem_formatted_command = rsem_command_string.format(
gtf_file=job_context["gtf_file_path"],
genes_to_transcripts=job_context["genes_to_transcripts_path"],
fasta_file=job_context["fasta_file_path"],
rsem_prefix=rsem_prefix
)
rsem_completed_command = subprocess.run(rsem_formatted_command.split())
salmon_command_string = ("salmon --no-version-check index -t {rsem_transcripts}"
" -i {index_dir} --type quasi -k {kmer_size}")
# This is the property we added in the Surveyor System section
# to denote whether this index is for long or short average read lengths.
kmer_size_property = BatchKeyValue.objects.get(batch=job_context["batches"][0],
key="kmer_size")
# rsem-prepare-reference outputs a transcripts.fa file which needs
# to be passed into salmon.
rsem_transcripts = rsem_prefix + ".transcripts.fa"
salmon_formatted_command = salmon_command_string.format(
rsem_transcripts=rsem_transcripts,
index_dir=job_context["output_dir"],
kmer_size=kmer_size_property.value)
salmon_completed_command = subprocess.run(salmon_formatted_command.split())
return job_context
It’s a big function, perhaps even a bit too big, but it really boils down to two shell calls. The first shell call to rsem-prepare-referenceoutputs the rsem_transcripts file which is then passed into salmon index. salmon-index outputs an entire directory which we want all of.
The next call in the pipeline, _zip_index(), takes care of zipping the entire directory into a single tarball along with setting job_context["files_to_upload"] = [job_context["gtf_file"]]. The reason for setting that property is that utils.upload_processed_files will look for it and, if it’s present in job_context, will only upload the files in that list to Permanent Storage. The File.upload_processed_file() method will use the File’s processed_format property to determine which file should be uploaded. Therefore even though we don’t want to upload the gtf_file, that File object knows how to upload its processed file, which in this case is the tarball we create in _zip_index(). All that’s left in our pipeline is utils.cleanup_raw_files() which will clear the raw data out of Temporary Storage and utils.end_job() which will free up the local disk space for the next job and mark the job as a success.
6. Conclusion
Once the ProcessorJob has been marked as a success in the database, we’re done with our example! We’ve certainly skipped over some sticky parts such as dealing with the idiosyncrasies of Ensembl’s REST API or less interesting code such as failure logging and handling. However, the goal of this blog post was to illustrate how the different components of the system work together to actually get some data from The Internet™ and refine it. I also only barely mentioned the Foreman, which makes sure that when failure inevitably happens, jobs will be retried a couple times before an administrator of the system is notified.
I’ve also omitted a discussion of the Aggregator, but that’s mostly because it’s still being fully specced out. What is defined about it is that it will have a REST API which our various interfaces will use to search, aggregate, and download data stored in refine.bio’s compendium. We’re currently gathering user feedback to better define its functionality. We’re also currently working on finding additional team members to help us implement it.
TL;DR: We need to put together lots of data from disparate sources at low cost via cloud workflows, and this technical post describes the CCDL’s system using specific examples. The refinery runs on AWS and uses Docker and Nomad. Its source code is available on GitHub.
Table of Contents
- Introduction
- The Surveyor System
- The Message System
- The Downloaders System
- The Processors System
- Conclusion
1. Introduction
In refine.bio, Part 1 I explained the need for refine.bio along with many of the specific requirements it would need to meet. In this post I will explain the design I came up to meet those requirements, implementation details about it, and then examples that follow how data is surveyed, downloaded, and then processed.
The design of refine.bio is focused around two abstractions: batches and jobs. A batch is a singular unit of data to be processed. It could be a sample, a genome, or another entity. A job is a unit of work that the system does. There are three kinds of jobs:
- Survey Job: A job that discovers new batches.
- Downloader Job: A job that downloads the data for one or more batches.
- Processor Job: A job that modifies data associated with one or more batches.
The most natural way to discuss the design of refine.bio is in terms of these two abstractions because they represent the core functionality of the system: it refines (via jobs) data (represented by batches). Making these concepts reified entities enables the system to record what it has done and when. The batches table records what data is available, where it can be downloaded from, and any other information the system has about the data. The tables for the various jobs will record when the data was discovered, when it was downloaded, and what processing steps the system performed on it. This will enable a UI to be built exposing this information to users, which will provide them with the context necessary to trust the fidelity of the data.
To help explain how this all comes together I will walk through an example I recently implemented which built Salmon indices for every organism contained in Ensembl Genomes. Salmon is a tool for analyzing RNASeq data which we are using in refine.bio to quantify transcripts. In order to be able to use Salmon, you first need a Salmon index for your transcriptome. Each Salmon index is specific for one organism, so we decided to use refine.bio to preprocess all the indices ahead of time. The basic outline of what we did for each organism is:
- Download the organism’s genome from Ensembl.
- Clean up the genome annotation file.
- Use RSEM to build a custom transcriptome.
- Feed that custom transcriptome into the salmon index command.
We’ll use this diagram as a map to follow our example as the data goes from being stored in Ensembl to being a fully processed Salmon index. I will note here that the code examples used in this post have been stripped down as much as possible to make them easy to read and understand. Therefore almost all of the housekeeping and error handling parts of the code have been omitted along with many comments.
2. The Surveyor System
We’re going to start by looking at how the Surveyor pulls metadata from Ensembl, creates Batches, and then queues DownloaderJobs. The way to create a new Surveyor is to create an implementation of the abstract ExternalSourceSurveyor class. ExternalSourceSurveyorhas three methods which we will need to override:
- discover_batches() - Discovers Batches and add them to the surveyor’s list of Batches by calling theExternalSourceSurveyor.add_batch() method.
- determine_pipeline() - Specifies which Processor Pipeline should run on each Batch. This will be stored as the pipeline_required property on the batches table.
- group_batches() - Groups Batches together that should be downloaded together.
The way these methods will be used together is best demonstrated by looking at the ExternalSourceSurveyor.survey() method:
def survey(self):
self.discover_batches()
for group in self.group_batches():
self.queue_downloader_jobs(group)
So what happens there is that ExternalSourceSurveyor and classes which inherit from it have a property self.batches which is a list of Batches. The discover_batches() method is expected to populate that property via the add_batch() method. For each Batchdiscover_batches() discovers, it calls add_batch(). This function takes required fields for the Batch table, a list of Files associated with the Batch, and a dictionary of key-value pairs which are tacked onto the Batch.
So let’s take a look at what our example’s discover_batches() method looks like:
def discover_batches(self):
ensembl_division = (
SurveyJobKeyValue
.objects
.get(survey_job_id=self.survey_job.id,
key__exact="ensembl_division")
.value
)
r = requests.get(DIVISION_URL_TEMPLATE.format(division=ensembl_division))
# Yes I'm aware that specieses isn't a word. However, I need to
# distinguish between a singular species and multiple species.
specieses = r.json()
for species in specieses:
self._generate_batches(species)
To break down what’s going on: the division of Ensembl which we’re surveying in this particular job is retrieved, a list of species is pulled from Ensembl’s REST API and a Batch is generated for each species. There are quite a few idiosyncrasies when working with the collection of APIs and FTP servers which make up Ensembl Genomes, so I’m not going to delve into everything _generate_batches() does. However, it primarily builds the appropriate URL to download the Batch’s files, creates File objects for both (GTF and FASTA) file types, and cleans up some of the metadata returned by Ensembl’s REST API. However, the most important thing it does is call ExternalSourceSurveyor.add_batch() like so:
self.add_batch(platform_accession_code=platform_accession_code,
experiment_accession_code=url_builder.file_name_species.upper(),
organism_id=url_builder.taxonomy_id,
organism_name=url_builder.scientific_name,
experiment_title="NA",
release_date=current_time,
last_uploaded_date=current_time,
files=[fasta_file, gtf_file],
key_values=species)
add_batch() then takes care of preventing duplicate Batches, creating the Batch objects, setting the correct Processor Pipeline (by calling the user-defined determine_pipeline() method), and saving everything to the database. For this source we only run one Processor Pipeline so we simply return the correct enumerated value. Therefore the implementation of determine_pipeline() is a single line that looks like this:
def determine_pipeline(self, batch: Batch, key_values: Dict = {}) -> ProcessorPipeline:
return ProcessorPipeline.TRANSCRIPTOME_INDEX
At this point we’ve implemented our determine_pipeline() and discover_batches() methods, leaving only group_batches(). Now there’s one detail about the Salmon Indices we’re building that I haven’t yet mentioned: we need two for each organism. We need one that is built to handle samples with a long average transcript read length and one for a short average transcript read length. The way we do this is to have _generate_batches() actually generate two Batches which are identical except that we add a ”kmer_size” key to the key_values dictionary that we pass through to add_batch(). However, both Batches need the same files to run. This setup allows us to only download them once.
We’re able to do this by creating a single DownloaderJob for both Batches. This is exactly what the ExternalSourceSurveyor.group_batches() method is for. We can group the Batches based on the download URL of their Files like so:
def group_batches(self) -> List[List[Batch]]:
"""Groups batches based on the download URL of their first File."""
download_url_mapping = {}
for batch in self.batches:
download_url = batch.files[0].download_url
if download_url in download_url_mapping:
download_url_mapping[download_url].append(batch)
else:
download_url_mapping[download_url] = [batch]
return list(download_url_mapping.values())
If we look back at the ExternalSourceSurveyor.survey() method:
def survey(self):
self.discover_batches()
for group in self.group_batches():
self.queue_downloader_jobs(group)
It will now queue DownloaderJobs for each of the groups returned by the group_batches() method we defined. At this point the Surveyor we’ve implemented will:
- Discover what species are stored in Ensembl.
- Record metadata and create two Batches for each species.
- Queue DownloaderJobs for each group of Batches.
This covers the responsibilities of the Surveyor system. In just a little bit we’ll take a look at what the the Downloaders System does with the jobs we’ve sent it. But first we’ll take a look at how the different systems send messages to each other.
3. The Message System
The Message System of refine.bio allows different components to communicate with each other. In the last section I mentioned that the Surveyor System queues DownloaderJobs, but I didn’t define what that meant. Queuing a DownloaderJob has two parts to it. The first creates a database entry representing the DownloaderJob. This entry records information about the job such as when it started, when it ended, and if it was successful. This allows the Foreman to check up on jobs to see if they need to be requeued and is preserved in case it is ever needed to verify the fidelity of a particular Batch of data.
The second part of queuing a DownloaderJob is to send a message to the Downloaders System so it knows that it has work to do. This is how both of these things happen:
@retry(stop_max_attempt_number=3)
def queue_downloader_jobs(self, batches: List[Batch]):
downloader_job = DownloaderJob.create_job_and_relationships(
batches=batches, downloader_task=self.downloader_task())
try:
send_job(downloader_task, downloader_job.id)
except:
# If the task doesn't get sent we don't want the
# downloader_job to be left floating
downloader_job.delete()
raise
The DownloaderJob.create_job_and_relationships() static method creates and saves a DownloaderJob to the database, along with creating links to each of the Batches it should process. Once that is done we send a message to the Downloaders System through the Message System via send_job(). If a failure occurs for any reason, such as a network partition, we delete the DownloaderJob so it’s not left in the database without a message telling the Downloaders System to pick it up. This entire method is wrapped with the decorator @retry(stop_max_attempt_number=3) to attempt to overcome temporary failures.
So, we send a message through the Message System to tell the Downloaders System to pick up the job, but how does that actually work? The Message System is not actually something I’ve built because distributed message queues are a hard problem that a lot of other people have expended a lot of effort to get right. There are a number of tools that could be used for the job such as RabbitMQ, Amazon SQS, or Apache Kafka. We use Hashicorp’s Nomad to manage messages.
Our decision to switch to Nomad was actually unrelated to the Message System. Nomad isn’t actually a message queue but it can be used as one for our purposes. We needed an orchestration tool to manage the cluster we’re going to use for the Downloaders System and the Processors System. Nomad works by running an agent on each node of the cluster. This agent does two primary things: it communicates with the rest of the cluster and it performs fingerprinting on the node. This fingerprinting discovers how many resources the node has available to it such as RAM, disk space, and CPU power. This allows the leader nodes to receive a job message and find a node with the available resources needed to run that job, whether it’s a DownloaderJob or a ProcessorJob. By carefully estimating the amount of resources each job will require, we efficiently use the cluster’s computing power. Nomad’s scheduling system then aims to solve a form of the bin packing problem where jobs are objects with different volumes and the nodes are the bins we want to pack as tightly as possible.
So when I discuss sending a message to the Message System what actually happens is that a Nomad job message is sent to the Nomad leaders, which then find an appropriate node of the cluster to perform the work. However, most contributors to the project won’t have to think too much about what is happening under the hood; they can just send a message to the Message System and trust it to find a node to perform the work associated with that job. So now that we know how the message that our Surveyor sends manages to make it to the Downloaders System we can dive into what that system does.
4. The Downloaders System
The Downloaders System is responsible for downloading the data for Batches from external sources. Once the data is downloaded it will be stored in the Temporary Storage, which we’re using AWS S3 for. When it comes time to process the data the Processors System, pulls it back out of Temporary Storage. It may seem inefficient to download the data, push it to Temporary Storage, and then pull it back out to process it. Why not just download it and then immediately process it without that other step? There are a couple reasons for this.
If the ProcessorJob fails for any reason we won’t have to re-download the data. Once a DownloaderJob has completed successfully we won’t need to re-download the data from the external source, no matter what issues arise while processing it. Also, this lets us use resources more efficiently. This ties into what I discussed in the last section about how Nomad’s fingerprinting and scheduling capabilities can be used to achieve efficient bin packing. DownloaderJobs have very low resource requirements but can take a while because we can only download data as fast as external servers will provide the data. ProcessorJobs on the other hand tend to have much higher resource requirements because of the processing work we’re performing. If we did not separate these two responsibilities then our MegaJobswould need to be allocated a large number of resources, which they would then not utilize for a long time while downloading the data from external sources. By having ProcessorJobs download the data from within the system, we get the benefit of the good network latency between AWS EC2 and S3.
With your burning questions of “why split those responsibilities?!?!?” answered, we can move on to what a Downloader for our example looks like. I am going to make use of the functions _download_file(download_url: str, file_path: str, job: DownloaderJob) and _upload_files(job_dir: str, files: List[File], job: DownloaderJob) without showing the code for them. This is because they pretty much just do what their names imply; most of their actual implementation involves handling and logging network errors. So here’s the main function for the Downloader:
def download_transcriptome(job_id: int) -> None:
# Block One
job = utils.start_job(job_id)
batches = job.batches.all()
# Prevents different jobs from using the same directory
job_dir = "downloader_job_" + str(job_id)
# Block Two
first_fasta_file = File.objects.get(batch=batches[0], raw_format__exact="fa.gz")
first_gtf_file = File.objects.get(batch=batches[0], raw_format__exact="gtf.gz")
second_fasta_file = File.objects.get(batch=batches[1], raw_format__exact="fa.gz")
second_gtf_file = File.objects.get(batch=batches[1], raw_format__exact="gtf.gz")
os.makedirs(first_fasta_file.get_temp_dir(job_dir), exist_ok=True)
# The two Batches share the same fasta and gtf files, so
# only download each one once
_download_file(first_fasta_file.download_url,
first_fasta_file.get_temp_pre_path(job_dir),
job)
_download_file(first_gtf_file.download_url,
first_gtf_file.get_temp_pre_path(job_dir),
job)
# Block Three
# Then create symlinks so the files for the second Batch
# can be found where they will be expected to.
os.symlink(first_fasta_file.get_temp_pre_path(job_dir),
second_fasta_file.get_temp_pre_path(job_dir))
os.symlink(first_gtf_file.get_temp_pre_path(job_dir),
second_gtf_file.get_temp_pre_path(job_dir))
_upload_files(job_dir,
[first_fasta_file, first_gtf_file, second_fasta_file, second_gtf_file],
job)
utils.end_job(job, batches, success=True)
This function is pretty long and there’re a few different parts so I’ve stuck in a few comments denoting different code blocks. Block One is just getting the job started. It calls utils.start_job() which will mark the job as started and retrieve the DownloaderJob object from the database. We then retrieve the Batches from the database and build our job_dir_prefix which we will put in every path we use during this job so we don’t overlap with any other jobs running concurrently on the same instance. In Block Two we retrieve the File objects out of the database, create a temporary directory to download files into, and then we actually download one set of files.
In Block Three we deal with the fact that we need the same files for two different batches. This goes back to what I discussed in the Surveyor System section about building two Salmon Indices for each organism. Rather than just re-download the exact same files again, we create symlinks to the locations that those files are expected to have been downloaded to. That’s what the File.get_temp_pre_path() method does, it returns where the path where the unprocessed file should be located (pre as in pre-processing). Now when we call the _upload_files() function it will have each of the Files we passed into it upload themselves. That’s right, each File object is capable of uploading itself to Temporary Storage via its upload_raw_file() method. Finally we just call utils.end_job() to mark the job as completed successfully. (In the actual function we catch errors and call utils.end_job() with success=False when appropriate.)
The one other thing that utils.end_job() will take care of for us is queuing a ProcessorJob. This is very similar to the way a DownloaderJob was queued earlier, so I’ll just move along to…
5. The Processors System
You’ve made it to the good stuff! The Processors System puts the refine in refine.bio. The full range of types of processing that we actually perform is outside the scope of this blog post, but they are carefully considered by the project’s Scientific Lead, Dr. Jaclyn Taroni. Instead we’ll focus on just the example we’ve been following throughout this post. The basic flow for all Processors is to retrieve the Batch’s data from Temporary Storage, perform some kind of processing, and then push the processed data into Permanent Storage. The processing we’ll need to perform for our example are the steps I outlined in the beginning of the post:
- Clean the genome annotation file.
- Use RSEM to build an custom transcriptome.
- Feed that custom transcriptome into the salmon index command.
However, before we can dive into actually doing all of that we need to discuss what Processors are. A Processor is actually just a function that accepts a single argument: the ID of a ProcessorJob. When the Processor System receives a job message from the Message System it will extract the ProcessorJob ID from the message along with the name of the Processor to execute. It then runs the function and waits for it to complete so it can report that the job’s computational resources can be used by other jobs.
Despite the minimal specification of Processors, there are certain responsibilities they are expected to complete. Each Processor must mark when it starts the job, when it ends the job, and whether or not the job was successful. They should also free up any disk space they utilize and if they’re successful they should remove the raw data from Temporary Storage. Since every Processor needs to do this stuff a framework is provided by the system to make it easy and to keep things DRY. The framework was designed with the goal of allowing reusable and composable functions to be created which can then be joined together into a single pipeline. Each function in the pipeline takes a job_context dictionary as an input and must output another (or the same) dictionary which can be passed into the next function of the pipeline. This behavior can be utilized by calling processors.utils.run_pipeline(), which takes two arguments: an initial job_context dictionary and a list of functions.
To illustrate this let’s take a look at the Processor function for our example:
def build_index(job_id: int) -> None:
utils.run_pipeline({"job_id": job_id},
[utils.start_job,
_set_job_prefix,
_prepare_files,
_process_gtf,
_create_index,
_zip_index,
utils.upload_processed_files,
utils.cleanup_raw_files,
utils.end_job])
Any function which returns a job_context containing ”success”: False will cause the pipeline to terminate and the job to be marked as a failure. Notice that the list of functions is composed of calls to functions defined in the utils namespace and private functions from the current namespace. The functions defined in the utils namespace take care of the common functionality I discussed in the previous paragraph. They pretty much all do what their names imply, but it will be helpful to note that in addition to marking the ProcessorJob as started, utils.start_job() also retrieves the ProcessorJob and Batch objects from the database and adds them to the job_context. So now let’s take a look at our pipeline step by step starting with:
def _set_job_prefix(job_context: Dict) -> Dict:
job_context["job_dir_prefix"] = "processor_job_" + str(job_context["job_id"])
return job_context
This adds the key “job_dir_prefix” to the job_context, which serves a similar purpose to the variable named similarly from the Downloaders System section: it makes all paths related to this job unique on the local disk. Next we run _prepare_files() which has the purpose of pulling the files from Temporary Storage and unzipping them so they’re ready to be processed.
def _prepare_files(job_context: Dict) -> Dict:
# Transcriptome index processor jobs have only one batch. Each
# batch has a fasta file and a gtf file
batch = job_context["batches"][0]
fasta_file = File.objects.get(batch=batch, raw_format__exact="fa.gz")
gtf_file = File.objects.get(batch=batch, raw_format__exact="gtf.gz")
fasta_file.download_raw_file(job_context["job_dir_prefix"])
gtf_file.download_raw_file(job_context["job_dir_prefix"])
gzipped_fasta_file_path = fasta_file.get_temp_pre_path(job_context["job_dir_prefix"])
gzipped_gtf_file_path = gtf_file.get_temp_pre_path(job_context["job_dir_prefix"])
job_context["fasta_file_path"] = gzipped_fasta_file_path.replace(".gz", "")
job_context["gtf_file_path"] = gzipped_gtf_file_path.replace(".gz", "")
with gzip.open(gzipped_fasta_file_path, "rb") as gzipped_file, \
open(job_context["fasta_file_path"], "wb") as gunzipped_file:
shutil.copyfileobj(gzipped_file, gunzipped_file)
with gzip.open(gzipped_gtf_file_path, "rb") as gzipped_file, \
open(job_context["gtf_file_path"], "wb") as gunzipped_file:
shutil.copyfileobj(gzipped_file, gunzipped_file)
job_context["fasta_file"] = fasta_file
job_context["gtf_file"] = gtf_file
return job_context
With our files unzipped locally, we’re ready to start processing by calling process_gtf(). This function cleans the genome annotation file (the gtf_file in this case) and creates a mapping from genes to transcripts for the given genome. If you’re not sure what that means, don’t worry about it; the upshot is that job_context["gtf_file_path"] is updated to point to the cleaned GTF file and the ”genes_to_transcripts_path” key is added to the job_context. Now we’re ready to create a Salmon Index using RSEM and Salmon:
def _create_index(job_context: Dict) -> Dict:
# Prepare the output directories.
work_dir = job_context["gtf_file"].get_temp_dir(job_context["job_dir_prefix"])
job_context["output_dir"] = os.path.join(work_dir, "index")
rsem_index_dir = os.path.join(work_dir, "rsem_index")
os.makedirs(rsem_index_dir, exist_ok=True)
# RSEM takes a prefix path and then all files generated by it will
# start with that
rsem_prefix = os.path.join(rsem_index_dir,
job_context["fasta_file"].get_base_name())
rsem_command_string = (
"rsem-prepare-reference --gtf {gtf_file}"
" --transcript-to-gene-map {genes_to_transcripts} {fasta_file} {rsem_prefix}"
)
rsem_formatted_command = rsem_command_string.format(
gtf_file=job_context["gtf_file_path"],
genes_to_transcripts=job_context["genes_to_transcripts_path"],
fasta_file=job_context["fasta_file_path"],
rsem_prefix=rsem_prefix
)
rsem_completed_command = subprocess.run(rsem_formatted_command.split())
salmon_command_string = ("salmon --no-version-check index -t {rsem_transcripts}"
" -i {index_dir} --type quasi -k {kmer_size}")
# This is the property we added in the Surveyor System section
# to denote whether this index is for long or short average read lengths.
kmer_size_property = BatchKeyValue.objects.get(batch=job_context["batches"][0],
key="kmer_size")
# rsem-prepare-reference outputs a transcripts.fa file which needs
# to be passed into salmon.
rsem_transcripts = rsem_prefix + ".transcripts.fa"
salmon_formatted_command = salmon_command_string.format(
rsem_transcripts=rsem_transcripts,
index_dir=job_context["output_dir"],
kmer_size=kmer_size_property.value)
salmon_completed_command = subprocess.run(salmon_formatted_command.split())
return job_context
It’s a big function, perhaps even a bit too big, but it really boils down to two shell calls. The first shell call to rsem-prepare-referenceoutputs the rsem_transcripts file which is then passed into salmon index. salmon-index outputs an entire directory which we want all of.
The next call in the pipeline, _zip_index(), takes care of zipping the entire directory into a single tarball along with setting job_context["files_to_upload"] = [job_context["gtf_file"]]. The reason for setting that property is that utils.upload_processed_files will look for it and, if it’s present in job_context, will only upload the files in that list to Permanent Storage. The File.upload_processed_file() method will use the File’s processed_format property to determine which file should be uploaded. Therefore even though we don’t want to upload the gtf_file, that File object knows how to upload its processed file, which in this case is the tarball we create in _zip_index(). All that’s left in our pipeline is utils.cleanup_raw_files() which will clear the raw data out of Temporary Storage and utils.end_job() which will free up the local disk space for the next job and mark the job as a success.
6. Conclusion
Once the ProcessorJob has been marked as a success in the database, we’re done with our example! We’ve certainly skipped over some sticky parts such as dealing with the idiosyncrasies of Ensembl’s REST API or less interesting code such as failure logging and handling. However, the goal of this blog post was to illustrate how the different components of the system work together to actually get some data from The Internet™ and refine it. I also only barely mentioned the Foreman, which makes sure that when failure inevitably happens, jobs will be retried a couple times before an administrator of the system is notified.
I’ve also omitted a discussion of the Aggregator, but that’s mostly because it’s still being fully specced out. What is defined about it is that it will have a REST API which our various interfaces will use to search, aggregate, and download data stored in refine.bio’s compendium. We’re currently gathering user feedback to better define its functionality. We’re also currently working on finding additional team members to help us implement it.
Related Post
Are you attending the American Association for Cancer Research (AACR) Annual Meeting in San Diego, CA? Visit us in the exhibit hall at booth 2918 from April 19-22, 2026, and during poster sessions. Meet Data Lab team members and learn about ALSF grant opportunities, resources for pediatric cancer research, and apply to free data science training workshops!
Applications are open for the Data Lab's upcoming workshop, which will cover advanced topics in the analysis of single-cell RNA-seq data for researchers studying pediatric cancer. The course will be held virtually from June 8-12, 2026 from 12-5pm Eastern time.
The Data Lab will be holding a virtual workshop, Introduction to Single-cell RNA-Sequencing, from May 11-15, 2026! In this workshop, Data Lab staff will introduce researchers studying pediatric cancer to the R programming language, the Tidyverse R packages for data science, single-cell RNA-seq data analysis, and annotating cell types.
Exciting news from the Single-cell Pediatric Cancer Atlas (ScPCA) Portal! All datasets on the portal have been updated to include several new features that enhance data quality and usability. Here’s a close look at what we’ve added and why.
It's that time of year again – it's time to start a new research project! Planning for the research itself can be daunting, but planning for your work to be fully reproducible is a whole other layer that isn't always emphasized. In this blog post, we'll outline a few project organization principles we teach in our Reproducible Research Practices workshop that we think are really helpful for ensuring reproducibility (among other benefits!).
At the Childhood Cancer Data Lab, we support pediatric cancer researchers by providing data science training, collaborating on data-intensive projects, and designing open-source tools. Through this work, we’ve had the opportunity to engage with scientists who are applying rigorous, data-driven approaches to address some of the most pressing challenges in childhood cancer.
Applications are open for the Data Lab's upcoming workshop, which will cover advanced topics in the analysis of single-cell RNA-seq data for researchers studying pediatric cancer. The course will be held virtually from December 8-12, 2025 from 12-5pm Eastern time.
At the Childhood Cancer Data Lab, we’re committed to helping pediatric cancer researchers work more efficiently, collaboratively, and reproducibly. That’s why we created our Reproducible Research Practices workshop, first piloted in 2022 with six participants. Since then, more than 50 pediatric cancer researchers have joined us to learn hands-on techniques for achieving reproducible results in computational cancer research. This fall, we’re excited to hold the next workshop in Philadelphia, PA!
The Data Lab recently traveled to California to lead a hands-on workshop for nine researchers from the UC Santa Cruz Treehouse Childhood Cancer Initiative. The participants, all from a range of backgrounds and experience levels, came together to learn common practices for reproducible computational research. Our relationship with Treehouse spans years, grounded in a shared commitment to open science and reproducibility. This workshop was a chance to strengthen that partnership and an opportunity to put shared values into practice!
Use cases define how users interact with a product or system, including actions users can take and how the system responds. It also identifies user goals and paths for the system to handle errors.
The Open Single-cell Pediatric Cancer Atlas (OpenScPCA) project is one year old, and there is much to celebrate! For the past year, we’ve worked closely with pediatric cancer experts to analyze data from the ScPCA Portal, improving its utility for researchers everywhere. Our focus has been on adding reliable cell type annotations across samples on the Portal, but the journey has been much more than that.
The Data Lab will be holding a virtual workshop, Introduction to Single-cell RNA-Sequencing, from August 4-8, 2025! In this workshop, Data Lab staff will introduce researchers studying pediatric cancer to the R programming language, the Tidyverse R packages for data science, single-cell RNA-seq data analysis, and annotating cell types.
Applications are open for the Data Lab's next training workshop! We will cover advanced topics in the analysis of single-cell RNA-seq data for researchers studying pediatric cancer. The 3-day course will take place June 10-12, 2025 from 9am-5pm Eastern time in Bala Cynwyd, PA, just outside of Philadelphia.
Before we launched OpenScPCA, we had to outline the process for contributing to analyses and then document that process for others. In addition, when designing the process for contributing to the project, we made sure to implement strategies to ensure reproducibility over the life cycle of the project. After planning and documenting expectations for contributors, we prepared to launch our first call for contributions, where we asked pediatric cancer experts to help us assign cell type annotations for all samples on the Portal. We thought it would be helpful to have an existing analysis module that other contributors could reference, so we picked a member of our science team (it’s me, hi 👋) to go through the process of developing an analysis module.
Are you attending the American Association for Cancer Research (AACR) Annual Meeting in Chicago, IL? Visit us in the exhibit hall at booth 3706 from April 27-30, 2025, and during poster sessions. We have exciting news about grant opportunities, projects, free training workshops, and more!
In 2023, we launched our first-ever call for contributions to the Single-cell Pediatric Cancer Atlas (ScPCA) Portal, inviting the research community to share their data. This initiative has been instrumental in expanding the Portal, with numerous pediatric cancer researchers responding to the call and collaborating with us to make more data available. Today, the Portal holds data from 700 samples across 55 cancer types, and we look forward to increasing those numbers with our latest call for contributions.
We are excited to announce that our next virtual workshop, Introduction to Single-cell RNA-Seq, will run from March 24-28,2025! In this workshop, Data Lab staff will introduce researchers studying pediatric cancer to the R programming language, the Tidyverse R packages for data science, single-cell RNA-seq data analysis, and annotating cell types.
Launching the Open Single-cell Pediatric Cancer Atlas (OpenScPCA) project in April 2024 was a highlight of our year! This community-driven initiative aims to analyze data from the ScPCA Portal, which currently holds 700 samples from over 55 pediatric cancer types. The project is a step forward in advancing our knowledge of pediatric cancers through single-cell analysis, and we're excited to expand OpenScPCA in 2025! To that end, we're reflecting on some of our recent accomplishments and how we can keep that momentum going into next year.
When the Data Lab launched the Single-cell Pediatric Cancer Atlas (ScPCA) Portal in 2022, we knew it was only the beginning! We started by making data easily available for the research community and received an overwhelmingly positive response. But we know firsthand from training hundreds of pediatric cancer researchers in analysis that making data available is just the first step. We’re increasing the impact of the Portal by listening to the growing ScPCA community. Now more researchers can contribute datasets, new features are continuously being developed, and we started an open, collaborative project to further explore the available data! Here’s a look back at how we’ve enhanced the ScPCA Portal in 2024.
In our last blog post, we shared some of the tools and methods we are using in the Open Single-cell Pediatric Cancer Atlas (OpenScPCA) project to ensure that the analysis code remains usable and runnable throughout the project. That post mainly focused on some of the most dynamic phases of the project, when contributors are adding new analysis modules and updating existing ones with more refined results. Here, we will discuss the test data that enables the methods and our approach to running the full set of analyses on real data.
Applications are open for the Data Lab's next training workshop! We will cover advanced topics in the analysis of single-cell RNA-seq data for researchers studying pediatric cancer. The 3-day course will take place December 10-12, 2024 from 9am-5pm Eastern time in Bala Cynwyd, PA, just outside of Philadelphia.
Earlier this year, we launched the Open Single-cell Pediatric Cancer Atlas (OpenScPCA) project, a collaborative project to openly analyze the data in the Single-cell Pediatric Cancer Atlas Portal on GitHub. We hope this project will bring transparently and expertly assigned cell type labels to the data in the Portal, help the community understand the strengths and limitations of applying existing single-cell methods to pediatric cancer data, and, frankly, allow us to meet more scientists in our community working with single-cell data (maybe you? 😄).
Recently, the Data Lab packed up and headed to the University of Minnesota (UMN) to host a workshop for 19 researchers. Participants with a variety of skill levels and backgrounds joined us from UMN, St. Jude Children’s Research Hospital, the Mayo Clinic, and the Medical University of South Carolina.
Applications are open for the Data Lab's next workshop! We will hold a Reproducible Research Practices Course on October 23-24, 2024 in Milwaukee, WI. Instructors will introduce principles and techniques to achieve reproducible results in computational cancer research. We’ll show you the fundamentals of commonly used approaches in reproducibility that you can apply to increase the impact of your research by making your findings more robust and reliable! To ensure that workshop attendees have a great hands-on experience, a very limited number of seats will be available.
We are excited to announce our next workshop, Introduction to Bulk RNA-Sequencing and Reproducible Research Practices, will take place in Minneapolis, MN from August 19-22, 2024! In this workshop, Data Lab staff will introduce researchers studying pediatric cancer to the R programming language, the Tidyverse R packages for data science, bulk RNA-seq data analysis, pathway analyses, and techniques to achieve reproducible results in computational cancer research.
In April 2024, we announced the Open Single-cell Pediatric Cancer Atlas (OpenScPCA) project. Since then, we’ve been working to build a supportive community while getting started on a few analysis ideas! We’re excited to see growing interest in the project, and we have some big news for prospective collaborators.
So you recently did some single-cell RNA sequencing and are working on analyzing your data. You’ve already quantified the gene expression data, performed any filtering, and normalized your data, but now what? You know you want to perform differential expression analysis or that you need to annotate the cell types found in your data, but there are so many different tools and methods for performing these analyses. How do you know which one is the best method for your dataset? Don’t worry, we’ve all been there – even experts in the single-cell field have been there.
The Open Single-cell Pediatric Cancer Atlas (OpenScPCA) is an open, collaborative project to analyze data from the Single-cell Pediatric Cancer Atlas (ScPCA) Portal, which currently holds over 500 samples from over 50 pediatric cancer types. OpenScPCA uses an open contribution model designed to allow experts worldwide to contribute and rapidly share the results of analyses in real time. The project was officially launched in April 2024.
In March 2022, we launched the Single-cell Pediatric Cancer Atlas (ScPCA) Portal to make uniformly processed single-cell and single-nuclei RNA-Seq data widely available to the childhood cancer research community. Initially, all data available on the Portal was generated through grants funded by Alex’s Lemonade Stand Foundation (ALSF) as part of the ScPCA project. But enabling access to ALSF-funded data was just the beginning of our vision.Sharing is key to ensuring the Portal’s continued growth. Our sights were set on allowing more pediatric cancer researchers to contribute data to the ScPCA Portal.
The Data Lab has just launched the brand new Open Single-cell Pediatric Cancer Atlas (OpenScPCA) project! This open, collaborative project aims to analyze data from the ScPCA Portal, which currently holds 500 samples from over 50 pediatric cancer types. We are seeking contributors with diverse skills and expertise to join the project!
We are excited to announce that our next virtual workshop, Introduction to Single-cell RNA-Seq, will run from June 10-14, 2024! In this workshop, Data Lab staff will introduce researchers studying pediatric cancer to the R programming language, the Tidyverse R packages for data science, single-cell RNA-seq data analysis, and annotating cell types.
Applications are open for the Data Lab's next workshop! We are holding a two-day course on Reproducible Research Practices and the Open Single-cell Pediatric Cancer Atlas (OpenScPCA) project from May 14-15, 2024. Please note that the OpenScPCA module is an optional part of the workshop. The course begins with an introduction to principles and techniques to achieve reproducible results in computational cancer research. On day two, you can choose to continue the workshop and learn how to put your skills to use for OpenScPCA, our new pediatric cancer research project.
Are you attending the American Association for Cancer Research (AACR) annual meeting in San Diego, CA? Visit the Alex’s Lemonade Stand Foundation (ALSF) Grants and Data Lab teams at booth 3755 in the exhibit hall from April 7-10 and during poster sessions on April 8. We will announce a new collaborative project and share exciting news about the Single-cell Pediatric Cancer Atlas Portal and training opportunities!
Did you know that 70% of the Alex’s Lemonade Stand Foundation (ALSF) Childhood Cancer Data Lab team are currently women? Advancing our mission to empower childhood cancer researchers with knowledge, data, and tools would not be possible without their expertise. On the International Day of Women and Girls in Science, we are excited to introduce you to these women who integrate science, engineering, and design to tackle some of the greatest challenges faced by the pediatric cancer research community!
I have a confession to make: I am lazy. Ok, maybe that's too strong. Let's go for a euphemism instead: I am efficient. I love learning handy tricks that make my life easier and make my job smoother with fewer hiccups along the way. This is one part of why, here in the Data Lab, we love automation - why waste our time on rote, repetitive, housekeeping tasks when we can get the bots to do it for us? In this blog post, we'll highlight a few tips about how you can use RStudio to code more efficiently.
Writing source code is a significant part of data-intensive biomedical research. Everything from cleaning and pre-processing data to generating publication figures can be accomplished programmatically. Increasingly, funding agencies and journals require researchers to share their code. To pick a few examples, the Data Lab’s parent organization, Alex’s Lemonade Stand Foundation (ALSF), has such a requirement for awardees, and PLoS Computational Biology requires authors to make code underlying results and conclusions available.
There is an old joke in computer science about how there are only two hard things: cache invalidation, naming things, and off-by-one errors. I’ll leave aside the first one as beyond my own expertise, but the second comes up all the time in my work as a biological data scientist. Naming variables and functions in my code is a constant struggle, but one I have to deal with on my own or with my team. Much bigger problems come up when trying to deal with all the various ways that people across the world use names when talking about the diseases they work on, the types of cells they are looking at, the experimental methods they are using, and just about every other aspect of their studies.
Applications are open for the Data Lab's next workshop! We will be holding a Reproducible Research Practices Course in-person on October 24-25, 2023. Instructors will introduce principles and techniques to achieve reproducible results in computational cancer research. We’ll show you the fundamentals of commonly-used approaches in reproducibility that you can apply to increase the impact of your research by making your findings more robust and reliable! To ensure that workshop attendees have a great hands-on experience, there will be a very limited number of seats available.
At the Center for Data-Driven Discovery in Biomedicine (D3b), I lead the Bioinformatics Translational Pediatric Oncology Team, a team of bioinformatics scientists. Our mission is to advance pediatric oncology research and precision medicine through collaboration and development of open-source analytical tools, frameworks, and data resources. In 1998, I lost my four year old cousin John Matthew to a brain tumor we now know was likely a diffuse intrinsic pontine glioma. So, it was bittersweet for me to see the Open Pediatric Brain Tumor Atlas (OpenPBTA) manuscript published in Cell Genomics on the last day of brain tumor awareness month this past year. But let’s rewind.
Writing effective documentation is challenging. Users might not always read every word in the documentation. They might even just scroll past large chunks of text, but we can accommodate those behaviors by structuring and formatting content appropriately.
In 2019, Alex’s Lemonade Stand Foundation (ALSF) established the Single-cell Pediatric Cancer Atlas (ScPCA) through awards for data generation and to create an atlas of single-cell gene expression profiles of pediatric cancers of different types and from different organ sites. The Data Lab launched the ScPCA Portal in 2022 to make uniformly processed, summarized single-cell and single-nuclei RNA-seq data and de-identified metadata available for download. The ScPCA Portal also supports other data modalities, such as bulk RNA-seq, CITE-seq, and spatial transcriptomics. The ScPCA Portal currently hosts data for over 500 pediatric tumor and patient-derived xenograft samples from more than 50 cancer types, and continues to grow. The Data Lab is seeking contributions to the ScPCA Portal from researchers with existing single-cell datasets.
We are excited to announce that our next workshop, Introduction to Single-cell RNA-Seq, will take place in-person from June 13-15, 2023! Data Lab staff will introduce researchers studying pediatric cancer to the R programming language, the Tidyverse R packages for data science, single-cell RNA-seq data analysis, annotating cell types, and more. The 3-day course will take place from 9am-5pm Eastern time in Bala Cynwyd, PA, just outside of Philadelphia. Travel reimbursement (up to a certain amount) is available for qualifying participants.
The Childhood Cancer Data Lab maintains a collection of uniformly processed single-cell data from pediatric cancer clinical samples and xenografts in the Single-cell Pediatric Cancer Atlas (ScPCA) Portal. Although access to preprocessed data saves researchers time, we know that the downloads from the ScPCA Portal are only the starting point. That’s why we’ve created downstream analysis workflows for commonly performed analyses. Instead of writing code wholesale, you can analyze data once you’ve configured these workflows.
We are excited to announce that our next virtual workshop, Introduction to Single-cell RNA-Seq, will run from May 15-19, 2023! In this workshop, Data Lab staff will introduce researchers studying pediatric cancer to the R programming language, the Tidyverse R packages for data science, single-cell RNA-seq data analysis, and annotating cell types.
Last year, the Data Lab launched the Single-cell Pediatric Cancer Atlas (ScPCA) Portal, which today holds uniformly processed single-cell gene expression data obtained from 8 separate labs, over 480 samples, and representing 38 cancer types. The portal is still growing as we continue to receive and process raw data from ScPCA investigators! All uniformly processed data is made available for download on the ScPCA Portal, giving researchers easy access to a growing database of summarized gene expression data and metadata to utilize for their own research. But how exactly did we make sure that all of the data was uniformly processed? And how are we able to ensure uniform processing for incoming samples as the portal continues to grow?
Are you attending the American Association for Cancer Research (AACR) annual meeting in Orlando, FL this year? Visit Alex's Lemonade Stand Foundation (ALSF) at booth 369 in the exhibit hall from April 16-19! You'll find information about ALSF's grants program, the Childhood Cancer Data Lab and more. The Data Lab will also be holding office hours during select time slots.
The Data Lab is excited to announce that our next training workshop will be held virtually from March 13-17, 2023! During this workshop, we will cover advanced topics in the analysis of single-cell RNA-seq data for researchers studying pediatric cancer. The workshop will take place each day from 12-5pm Eastern. Each day consists of lectures and designated time for attendees to work on exercise materials and their own projects with our staff available for consultation. You’ll need a laptop with internet access and to install Zoom and Slack. You will log into an RStudio Server hosted by the Data Lab from your web browser. Pediatric cancer researchers are encouraged to apply now!
In September 2022, the Open Pediatric Brain Tumor Atlas (OpenPBTA) project culminated (for now) in a preprint on bioRxiv. This project, started in late 2019 and co-organized with the Center for Data Driven Discovery in Biomedicine (D3b) at Children’s Hospital of Philadelphia (CHOP), is a collaborative effort to comprehensively describe the Pediatric Brain Tumor Atlas (PBTA), a collection of multiple data types from tens of tumor types (read more about why crowdsourcing expertise for the study of pediatric brain tumors is important here). The project is designed to allow for contributions from experts across multiple institutions. We’ve conducted analysis and drafting of the manuscript openly on the version-control platform GitHub from the project’s inception to facilitate those contributions.
Recently, we told you about the Single-cell Pediatric Cancer Atlas (ScPCA) downstream analysis workflow. This ready-to-go workflow is intended to be used with single-cell and single-nuclei gene expression data available on the ScPCA Portal. We developed this workflow to filter, normalize, and perform dimensionality reduction, as well as incorporate initial clustering results to each processed sample/library object. Now we’re excited to introduce one of our latest offerings for use with ScPCA data, a clustering analysis workflow, which can be applied to datasets after running the filtering, normalization, and dimensionality reduction workflow!
The Data Lab is excited to announce that our next training workshop will be held in-person from January 31-February 2, 2023! During this workshop, we will cover advanced topics in the analysis of single-cell RNA-seq data for researchers studying pediatric cancer. The 3-day course will take place from 9am-5pm Eastern time in Bala Cynwyd, PA, just outside of Philadelphia. Travel reimbursement is available for qualifying participants.
Welcome to the Data Lab’s December Scientific Community Bulletin! Each month we share upcoming opportunities from Alex’s Lemonade Stand Foundation (ALSF), the Data Lab, and other events that we have gathered from a variety of science and research organizations. Subscribe to our blog to be alerted about future Scientific Community Bulletin posts!
In this blog post, I’d like to give an overview of the refine.bio refactoring process and web accessibility considerations. Through this process, our goal is to enhance the site usability and performance by improving the code quality and making the application more accessible. But before going into more details about them, let me provide you a quick history of refine.bio.
Welcome to the Data Lab’s November Scientific Community Bulletin! Each month we share upcoming opportunities from Alex’s Lemonade Stand Foundation (ALSF), the Data Lab, and other events that we have gathered from a variety of science and research organizations. Subscribe to our blog to be alerted about future Scientific Community Bulletin posts!
Here at the Data Lab, we're all about, well, data! We believe that data sharing and accessibility is key to accelerating the research process, and ultimately to improving outcomes for childhood cancer patients. So, we were excited to learn that one of the goals of the NCI/NIH initiative, the Childhood Cancer Data Initiative (CCDI), is to build up a Data Ecosystem that will facilitate pediatric cancer researchers' ability to explore and collect data from disparate resources. Although this Ecosystem is still in the early stages, several components are already being developed and are available for researchers to use! One component that is particularly interesting to us is the CCDI's Childhood Cancer Data Catalog (CCDC).
Welcome to the October Scientific Community Bulletin! Each month we share upcoming opportunities from Alex’s Lemonade Stand Foundation (ALSF), the Data Lab, and other events that we have gathered from a variety of science and research organizations. Subscribe to our blog to be alerted about future Scientific Community Bulletin posts!
Welcome to the September Scientific Community Bulletin! Each month we share upcoming opportunities from Alex’s Lemonade Stand Foundation (ALSF), the Data Lab, and other events that we have gathered from a variety of science and research organizations. Subscribe to our blog to be alerted about future Scientific Community Bulletin posts!
At the Data Lab, we are constantly looking for ways to enhance the tools we build for pediatric cancer researchers. Earlier this year, we launched the Single-cell Pediatric Cancer Atlas portal, a database of uniformly-processed single-cell data from pediatric cancer clinical samples. One way we felt the portal could be even more beneficial to pediatric cancer researchers is with a ready-to-go workflow that takes in single-cell data and prepares it for downstream analyses such as unsupervised clustering.
The Data Lab is excited to announce our next virtual workshop running from September 19-23, 2022! In this workshop, Data Lab staff will introduce researchers studying pediatric cancer to the R programming language, the Tidyverse R packages for data science, single-cell RNA-seq data analysis, and pathway analysis.
The Data Lab teaches data science courses targeted toward pediatric cancer researchers that introduce topics such as analysis of gene expression in bulk and single-cell data and principles of reproducible research. I wrote previously about how we use RStudio Server for our remote courses to simplify setup, and I wanted to write a bit more about some of the instructional practices we use so that our participants get the best experience we can provide. In particular, I wanted to talk about our use of live coding to facilitate active learning, and one of the tools we developed to make our course development just a bit easier.
Welcome to the August Scientific Community Bulletin! Each month we share upcoming opportunities from Alex’s Lemonade Stand Foundation (ALSF), the Data Lab, and other events that we have gathered from a variety of science and research organizations.
Often when building a server-client web application, we will encounter a situation where we want to send requests to our API in the chronological order that they occur on the client. Due to the asynchronous nature of these requests, it might not be possible to send them in the same callback for the event that triggered them. This is because we want to use the response from the previous request to craft our current one. A solution to this problem would be to implement a queue. Instead of calling the API immediately after events occur, implementing a queue ensures the latest data is sent with any request.
Welcome to the July Scientific Community Bulletin! Each month we share upcoming opportunities from Alex’s Lemonade Stand Foundation (ALSF), the Data Lab, and other events that we have gathered from a variety of science and research organizations. Subscribe to our blog to be alerted about future Scientific Community Bulletin posts!
Welcome to the Childhood Cancer Data Lab’s new blog feature, the monthly Scientific Community Bulletin! At the start of each month, we will share upcoming opportunities from Alex’s Lemonade Stand Foundation (ALSF), the Data Lab, and other events that we have gathered from a variety of science and research organizations. Our goal is to promote learning opportunities and highlight some of the excellent resources that our community provides.
At the Data Lab, our science team has a practice where an individual team member shares something that they recently figured out (or didn’t totally figure out yet) on a biweekly basis. We call this short 5-10 minute presentation How I Solved This, and it’s a great way to formally share (often hard-won) knowledge with each other. In this post, we thought we’d share how we solved something with the `renv` package with you.
The Childhood Cancer Data Lab builds resources guided by the most pressing needs of our primary users: pediatric cancer researchers. As the Data Lab's UX Designer, I conduct research activities with scientists like usability evaluations, semi-structured interviews, and card sorts to gain insight into their activities, processes, pain-points, and behaviors. I work with scientists and engineers at the Data Lab to use this information to improve existing products and services or to create new ones.
The Data Lab is excited to announce that our next training workshop is taking place in-person on Friday, June 10, 2022! During this full day workshop, instructors will introduce principles and techniques to achieve reproducible results in computational cancer research. We’ll show you the fundamentals of commonly-used approaches in reproducibility that you can apply to increase the impact of your research by making your findings more robust and reliable!
The Childhood Cancer Data Lab is growing as a resource for pediatric cancer researchers and we have more to offer to our community now, than ever before. Transitioning to our new and improved website is an exciting milestone, and here, we look forward to sharing progress, introducing new initiatives, and cultivating more opportunities to support childhood cancer research. Welcome to our new virtual home!
The Single-cell Pediatric Cancer Atlas (ScPCA) project began in 2019 when Alex’s Lemonade Stand Foundation (ALSF) funded 10 awards for single-cell profiling of pediatric cancer samples. The goal was to produce an atlas of gene expression profiles for a variety of childhood cancer types from different organ sites.
The CCDL team includes science, engineering, and design expertise. Combining these three disciplines in different ways across projects enables us to carry out our mission.
Here at the CCDL we value putting publicly available data to work. For example, we are currently processing and normalizing 1.5 million publicly available gene expression samples totaling ~$1.5 billion research dollars expended.
Like many teams that work with large amounts of external software, we run into issues with our transitive dependencies. In general, transitive dependencies are a hard problem to solve.
Though technology can introduce great benefit into our lives, it is often accompanied by a substantial amount of time and some expected frustration before we can reap the rewards. The time spent learning a new technology is what we usually call a learning curve.
The workshop will last from 9AM to 5PM on October 14th, 15th, and 16th at the CCDL offices at 1429 Walnut St Philadelphia, PA, 19102.
MultiPLIER is a machine learning approach that brings big data to bear on rare diseases. It’s also an example of the scientific approach and ethos of the CCDL, and the publication is a great opportunity to share how the CCDL is developing new technologies to accelerate research into cures for childhood cancers!
The Childhood Cancer Data Lab powered by Alex's Lemonade Stand Foundation is hosting a workshop to introduce childhood cancer researchers to reproducible analysis of bulk and single-cell transcriptomic data.
The Childhood Cancer Data Lab (CCDL), an initiative of Alex's Lemonade Stand Foundation develops tools, trainings, and methods to empower childhood cancer researchers. The work at the CCDL is focused and impactful. There are multiple opportunities and challenges for you to apply and grow your skills as a scientist or as an engineer.
The Childhood Cancer Data Lab powered by Alex's Lemonade Stand Foundation is hosting a workshop to introduce childhood cancer researchers to reproducible analysis of bulk and single-cell transcriptomic data.
At this hands-on, 3-day session held in Houston, researchers learned data science skills that could accelerate their own work. Drawing on skills learned at the workshop, childhood cancer researchers can perform basic analyses of their work to make informed decisions on how to proceed with their own research. Don’t just take our word for it, though. Read more about the workshop’s incredibly valuable benefits through its attendees’ perspectives.
The goal of our refine.bio project is to download, process, and make available gene expression datasets that can be analyzed together, or in parts, depending on a researcher’s need. Childhood cancer researchers need to be able to use data generated through multiple profiling technologies including microarrays and RNA-sequencing.
There are countless log blog posts out there about the benefits of good logging, how to log well, and how much to log. Going through them all can be a real log blog slog. Wouldn't it be cool if you could log like this:logger.info("Something happened!", job=job.id, user=user.id) and get an easily searchable output.
Caffeine is a stimulant that can induce alertness in certain individuals when consumed at an appropriate quantity. Caffeine is often obtained by ingesting caffeine-containing solutions. However, no protocol for obtaining caffeine from dehydrated, roasted beans using materials typically available in a Philadelphia office has been described in the published literature.
Alex’s Lemonade Stand Foundation (ALSF) staunchly believes that stronger scientific sharing practices will accelerate the pace of discovery and finding cures for children with cancer. Robust sharing improves reproducibility, minimizes redundant studies and maximizes our return on research investment.
Earlier this year, Alex’s Lemonade Stand Foundation identified single-cell gene expression profiling as an opportunity to build an atlas of cell types within tumors that could be broadly reused by pediatric cancer researchers.
This year was a big one for the CCDL. In our mission to empower pediatric cancer experts poised for big discoveries with the knowledge, data and methods to reach them we launched a software product, developed and delivered training workshops on single-cell and bulk RNA-seq analysis, and hired our data science team among other milestones.
I’m a scientist at Sage Bionetworks, a nonprofit research organization in Seattle, WA. My work focuses on a family of rare pediatric diseases (NF): neurofibromatosis type 1, type 2, and schwannomatosis.
Our particular process is designed to source opportunities from our team members and external stakeholders, convert those opportunities into a set of potential goals, and then select the goals that we expect will most advance our mission.
The ability to restore scroll position is often critical for website usability. It helps users keep the flow of navigation when going back and forth between different pages. Most modern browsers take care of restoring the scroll position automatically, but it doesn’t always work for Single Page Applications where the content is generated on the client’s side, often asynchronously.
Carnegie Mellon University Libraries is partnering with the Childhood Cancer Data Lab (CCDL), founded by Alex’s Lemonade Stand Foundation, to host a Data Analysis workshop using CCDL materials.
The CCDL will have a team of scientists at the American Association for Cancer Research 2020 Annual Meeting in sunny San Diego! Our team members are excited to talk to researchers studying pediatric cancer at Booth 1601.
To help keep pediatric cancer research moving forward, here are 3 ways the CCDL is helping the research community during this time: refine.bio, virtual workshops, and the Open Pediatric Brain Tumor Atlas project.
We know that pandemic-related university closures mean that the demand for opportunities for pediatric cancer researchers to increase their analytical skills has never been higher. As such, we are delighted to announce a pilot virtual workshop running from May 4-8, 2020!
Here at the Childhood Cancer Data Lab, we value transparency and the practice of open science. Much of the work we’ve done and the products that we build hinge on the generosity and openness of other scientists. In this post, as part of National Brain Tumor Awareness month, we want to talk about a project that our science team has been working on over the last few months (and to do so in a way that aligns with our values).
The workshop will take place on June 22 - 26, 2020 from noon - 5pm Eastern. Each day consists of lectures and designated time for attendees to work on exercise materials and their own projects with CCDL staff available for consultation.
When the CCDL (along with everyone else) realized that we would have to conduct our bioinformatics training workshops remotely, we had to make some quick decisions about how we were going to do it. Most of the instructional materials for our in person workshops were already online, so we knew we had a good base to work from. We just needed to figure how to adapt the live instruction.
At Alex’s Lemonade Stand Foundation’s Childhood Cancer Data Lab, we’re excited to be helping out with an upcoming event hosted by the Children’s Tumor Foundation. If you participate, you may meet members of our team who are mentoring and judging.
The workshop will take place on March 22 - 26, 2021 from noon - 5pm Eastern. Each day consists of lectures and designated time for attendees to work on exercise materials and their own projects with CCDL staff available for consultation
The workshop will take place on June 28- July 2, 2021 from noon to 5pm eastern. Each day consists of lectures and designated time for attendees to work on exercise materials and their own projects with CCDL staff available for consultation.
Hack4Rare is a virtual event that calls for healthcare startups, developers, solutions architects, and hackathon enthusiasts to join researchers, clinicians and patients in developing solutions built around a number of rare diseases including neurofibromatosis, PTEN Hamartoma Tumor Syndrome, RASopathies and Desmoid Tumors.
The workshop will take place on September 20 - 24, 2021 from noon - 5pm Eastern. Each day consists of lectures and designated time for attendees to work on exercise materials and their own projects with CCDL staff available for consultation.

333 E. Lancaster Ave, #414
Wynnewood, PA 19096 USA
© Childhood Cancer Data Lab