Does Bulk Tissue Still Belong in a Single-Cell Atlas?
Earlier this year, Alex’s Lemonade Stand Foundation identified single-cell gene expression profiling as an opportunity to build an atlas of cell types within tumors that could be broadly reused by pediatric cancer researchers. We in the Childhood Cancer Data Lab worked with the grant-making side of ALSF to design a Single-cell Pediatric Cancer Atlas RFA that would maximize the reusability of atlas datasets. One of the bullet points in the RFA that we’ve gotten a number of questions about is “applicants are required to perform RNA-sequencing of the bulk tissue from which the single cells are derived.” We wanted to take this opportunity to outline why we thought that including bulk sequencing would maximize reusability. N.B.: This post does not contain all available references on a topic: we selected a few from many possible references. Commenting is enabled on this post for the next month, so feel free to leave your favorite references on the topic as well.
Let’s briefly define what we mean by single-cell and bulk profiling. When we refer to bulk gene expression profiling, we’re discussing a process whereby RNA from throughout that tissue is profiled together. Bulk profiling can’t easily tell us what’s going on within each individual cell, but we do get to observe the extent to which each gene is expressed in that section of tissue. Single-cell profiling involves a step where bulk tissue is separated into individual cells, and then gene expression levels in those cells are measured separately for each cell.
Why build a single-cell pediatric cancer atlas?
While pediatric cancers are, collectively, the largest killer by disease of children in the US, they are each individually rare. This means that we couldn’t count on a future where each scientist would be able to rapidly build their own single-cell dataset from fresh tissues for each cancer type of interest. We needed a design that would allow researchers who have existing datasets, which were profiled from bulk tissue, to better understand their own data in the context of the single-cell atlas projects.
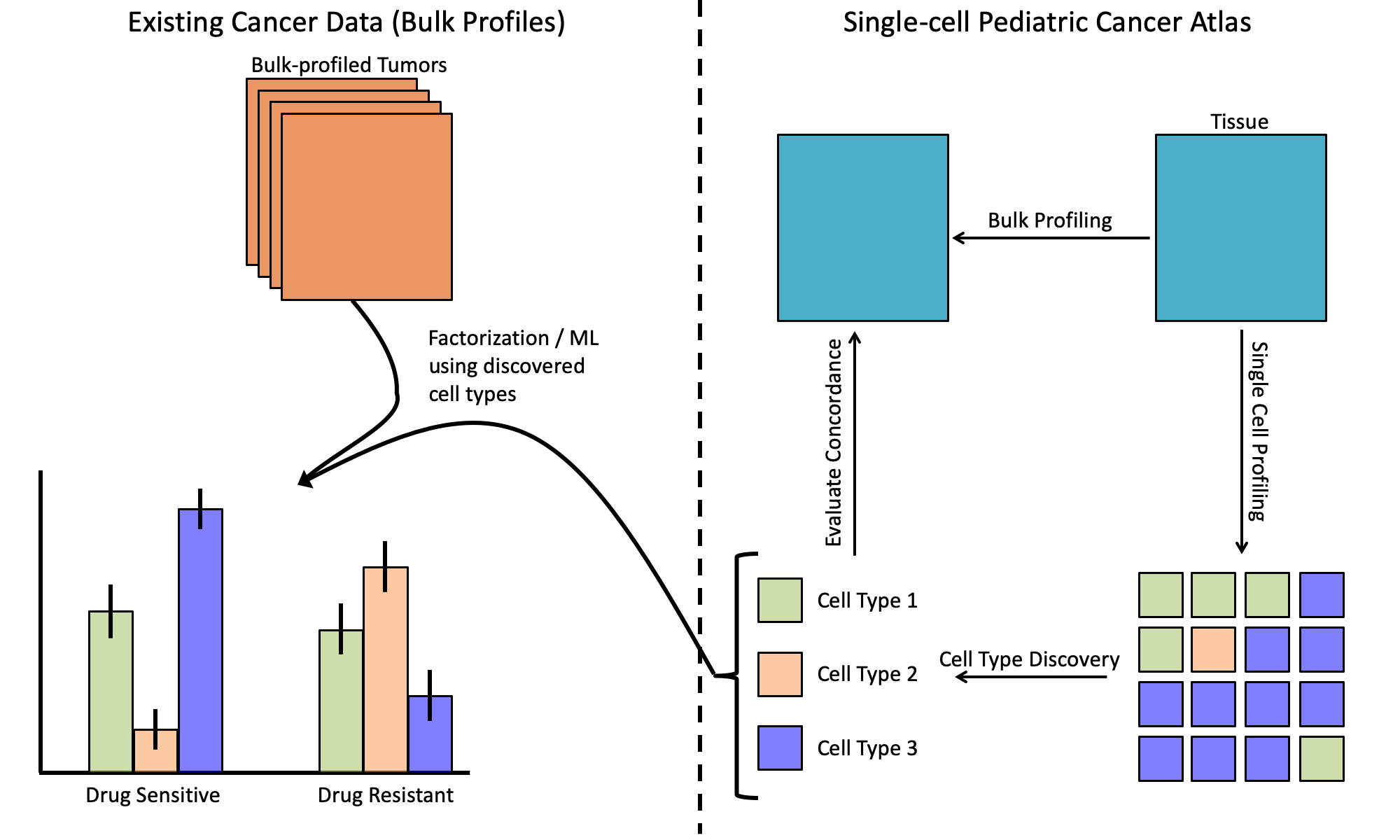
The arguments against requiring bulk profiling
- The coexpression levels of genes in bulk samples, especially of non-malignant tissue, is often driven by differences in cell-type composition. [Farahbod & Pavlidis]
- It’s possible to create pseudo-bulk data by aggregating data from single cells. [Ding et al. Sup Fig 10]
- There are deconvolution strategies that can use expression profiles from single-cell RNA-seq data to estimate the relative proportions of cell types in bulk-profiled samples. [Jew et al. among a growing number of others]
The arguments for requiring bulk profiling
- Dissociation is a physical process. Even if the pseudo-bulk profiles for certain tissue types display a high concordance with bulk, there is no guarantee that this concordance will still be high in these malignant tissues. For example, the data from Ding et al. focuses on normal tissue. If certain cell types are preferentially lost during dissociation then pseudo-bulk would be biased in different ways from traditional bulk measurement, which could make comparison difficult. Imagine that in the figure above, Cell Type 1 is lost during single-cell profiling. There is some risk that the atlas could fail to identify one or more cell types.
- Deconvolution strategies could still be helpful as long as cell-type reference profiles could be defined. However, this requires that the profiles of each cell type be available. Though this appears to often be the case for normal tissue. However, Suvà and Tirosh note in their recent review, “An emerging theme from recent scRNA-seq studies is that malignant cells tend to cluster in their expression profiles primarily by patient sample, and non-malignant cells cluster in their expression profiles by cell type, somewhat independently of the patient of origin (Filbin et al., 2018; Jerby-Arnon et al.,2018; Tirosh et al., 2016a, 2016b; Venteicher et al., 2017). This indicates that (1) inter-tumor heterogeneity is typically larger for malignant cells than for any particular type of non-malignant cells. (2) For malignant cells, inter-tumor heterogeneity is much larger than intra-tumor heterogeneity.” Imagine in the figure above that Cell Type 2’s expression differs greatly from patient-to-patient. There is some risk that deconvolution may be less successful if the inter-tumor expression heterogeneity for malignant cells is large for one or more of the profiled cancer types.
Why we asked for bulk profiling
On balance, there were enough unknown factors or areas of potential risk with a single-cell only approach for malignant tissue profiling that we elected to require bulk sequencing. As the RFA was being designed, it was not clear what the right approach would be for pediatric tumor tissues. These cancers are often somewhat biologically distinct from the adult cancers that much of the literature-to-date for single-cell profiling of human tumors has focused on. There have been a fair number of single-cell profiles of both PDX and mouse model samples, but it wasn’t clear if that data would be directly applicable to an atlas of human tumors.
Looking at the available literature and the uncertainties that existed, we thought it would be prudent to request candidates obtain bulk assays at the same time given the relative risks and benefits plus the modest cost of bulk sequencing relative to single-cell profiling. This means that we’ll be able to evaluate the concordance between single-cell profiles and what is observed in bulk samples. We hope that the references and literature can help others who are independently designing studies of malignancies, particularly pediatric ones, to best allocate resources towards their own efforts.
Earlier this year, Alex’s Lemonade Stand Foundation identified single-cell gene expression profiling as an opportunity to build an atlas of cell types within tumors that could be broadly reused by pediatric cancer researchers. We in the Childhood Cancer Data Lab worked with the grant-making side of ALSF to design a Single-cell Pediatric Cancer Atlas RFA that would maximize the reusability of atlas datasets. One of the bullet points in the RFA that we’ve gotten a number of questions about is “applicants are required to perform RNA-sequencing of the bulk tissue from which the single cells are derived.” We wanted to take this opportunity to outline why we thought that including bulk sequencing would maximize reusability. N.B.: This post does not contain all available references on a topic: we selected a few from many possible references. Commenting is enabled on this post for the next month, so feel free to leave your favorite references on the topic as well.
Let’s briefly define what we mean by single-cell and bulk profiling. When we refer to bulk gene expression profiling, we’re discussing a process whereby RNA from throughout that tissue is profiled together. Bulk profiling can’t easily tell us what’s going on within each individual cell, but we do get to observe the extent to which each gene is expressed in that section of tissue. Single-cell profiling involves a step where bulk tissue is separated into individual cells, and then gene expression levels in those cells are measured separately for each cell.
Why build a single-cell pediatric cancer atlas?
While pediatric cancers are, collectively, the largest killer by disease of children in the US, they are each individually rare. This means that we couldn’t count on a future where each scientist would be able to rapidly build their own single-cell dataset from fresh tissues for each cancer type of interest. We needed a design that would allow researchers who have existing datasets, which were profiled from bulk tissue, to better understand their own data in the context of the single-cell atlas projects.
The arguments against requiring bulk profiling
- The coexpression levels of genes in bulk samples, especially of non-malignant tissue, is often driven by differences in cell-type composition. [Farahbod & Pavlidis]
- It’s possible to create pseudo-bulk data by aggregating data from single cells. [Ding et al. Sup Fig 10]
- There are deconvolution strategies that can use expression profiles from single-cell RNA-seq data to estimate the relative proportions of cell types in bulk-profiled samples. [Jew et al. among a growing number of others]
The arguments for requiring bulk profiling
- Dissociation is a physical process. Even if the pseudo-bulk profiles for certain tissue types display a high concordance with bulk, there is no guarantee that this concordance will still be high in these malignant tissues. For example, the data from Ding et al. focuses on normal tissue. If certain cell types are preferentially lost during dissociation then pseudo-bulk would be biased in different ways from traditional bulk measurement, which could make comparison difficult. Imagine that in the figure above, Cell Type 1 is lost during single-cell profiling. There is some risk that the atlas could fail to identify one or more cell types.
- Deconvolution strategies could still be helpful as long as cell-type reference profiles could be defined. However, this requires that the profiles of each cell type be available. Though this appears to often be the case for normal tissue. However, Suvà and Tirosh note in their recent review, “An emerging theme from recent scRNA-seq studies is that malignant cells tend to cluster in their expression profiles primarily by patient sample, and non-malignant cells cluster in their expression profiles by cell type, somewhat independently of the patient of origin (Filbin et al., 2018; Jerby-Arnon et al.,2018; Tirosh et al., 2016a, 2016b; Venteicher et al., 2017). This indicates that (1) inter-tumor heterogeneity is typically larger for malignant cells than for any particular type of non-malignant cells. (2) For malignant cells, inter-tumor heterogeneity is much larger than intra-tumor heterogeneity.” Imagine in the figure above that Cell Type 2’s expression differs greatly from patient-to-patient. There is some risk that deconvolution may be less successful if the inter-tumor expression heterogeneity for malignant cells is large for one or more of the profiled cancer types.
Why we asked for bulk profiling
On balance, there were enough unknown factors or areas of potential risk with a single-cell only approach for malignant tissue profiling that we elected to require bulk sequencing. As the RFA was being designed, it was not clear what the right approach would be for pediatric tumor tissues. These cancers are often somewhat biologically distinct from the adult cancers that much of the literature-to-date for single-cell profiling of human tumors has focused on. There have been a fair number of single-cell profiles of both PDX and mouse model samples, but it wasn’t clear if that data would be directly applicable to an atlas of human tumors.
Looking at the available literature and the uncertainties that existed, we thought it would be prudent to request candidates obtain bulk assays at the same time given the relative risks and benefits plus the modest cost of bulk sequencing relative to single-cell profiling. This means that we’ll be able to evaluate the concordance between single-cell profiles and what is observed in bulk samples. We hope that the references and literature can help others who are independently designing studies of malignancies, particularly pediatric ones, to best allocate resources towards their own efforts.
Related Post
In March 2022, we launched the Single-cell Pediatric Cancer Atlas (ScPCA) Portal to make uniformly processed single-cell and single-nuclei RNA-Seq data widely available to the childhood cancer research community. Initially, all data available on the Portal was generated through grants funded by Alex’s Lemonade Stand Foundation (ALSF) as part of the ScPCA project. But enabling access to ALSF-funded data was just the beginning of our vision.Sharing is key to ensuring the Portal’s continued growth. Our sights were set on allowing more pediatric cancer researchers to contribute data to the ScPCA Portal.
The Data Lab has just launched the brand new Open Single-cell Pediatric Cancer Atlas (OpenScPCA) project! This open, collaborative project aims to analyze data from the ScPCA Portal, which currently holds 500 samples from over 50 pediatric cancer types. We are seeking contributors with diverse skills and expertise to join the project!
We are excited to announce that our next virtual workshop, Introduction to Single-cell RNA-Seq, will run from June 10-14, 2024! In this workshop, Data Lab staff will introduce researchers studying pediatric cancer to the R programming language, the Tidyverse R packages for data science, single-cell RNA-seq data analysis, and annotating cell types.
Applications are open for the Data Lab's next workshop! We are holding a two-day course on Reproducible Research Practices and the Open Single-cell Pediatric Cancer Atlas (OpenScPCA) project from May 14-15, 2024. Please note that the OpenScPCA module is an optional part of the workshop. The course begins with an introduction to principles and techniques to achieve reproducible results in computational cancer research. On day two, you can choose to continue the workshop and learn how to put your skills to use for OpenScPCA, our new pediatric cancer research project.
Are you attending the American Association for Cancer Research (AACR) annual meeting in San Diego, CA? Visit the Alex’s Lemonade Stand Foundation (ALSF) Grants and Data Lab teams at booth 3755 in the exhibit hall from April 7-10 and during poster sessions on April 8. We will announce a new collaborative project and share exciting news about the Single-cell Pediatric Cancer Atlas Portal and training opportunities!
Did you know that 70% of the Alex’s Lemonade Stand Foundation (ALSF) Childhood Cancer Data Lab team are currently women? Advancing our mission to empower childhood cancer researchers with knowledge, data, and tools would not be possible without their expertise. On the International Day of Women and Girls in Science, we are excited to introduce you to these women who integrate science, engineering, and design to tackle some of the greatest challenges faced by the pediatric cancer research community!
I have a confession to make: I am lazy. Ok, maybe that's too strong. Let's go for a euphemism instead: I am efficient. I love learning handy tricks that make my life easier and make my job smoother with fewer hiccups along the way. This is one part of why, here in the Data Lab, we love automation - why waste our time on rote, repetitive, housekeeping tasks when we can get the bots to do it for us? In this blog post, we'll highlight a few tips about how you can use RStudio to code more efficiently.
Writing source code is a significant part of data-intensive biomedical research. Everything from cleaning and pre-processing data to generating publication figures can be accomplished programmatically. Increasingly, funding agencies and journals require researchers to share their code. To pick a few examples, the Data Lab’s parent organization, Alex’s Lemonade Stand Foundation (ALSF), has such a requirement for awardees, and PLoS Computational Biology requires authors to make code underlying results and conclusions available.
There is an old joke in computer science about how there are only two hard things: cache invalidation, naming things, and off-by-one errors. I’ll leave aside the first one as beyond my own expertise, but the second comes up all the time in my work as a biological data scientist. Naming variables and functions in my code is a constant struggle, but one I have to deal with on my own or with my team. Much bigger problems come up when trying to deal with all the various ways that people across the world use names when talking about the diseases they work on, the types of cells they are looking at, the experimental methods they are using, and just about every other aspect of their studies.
Applications are open for the Data Lab's next workshop! We will be holding a Reproducible Research Practices Course in-person on October 24-25, 2023. Instructors will introduce principles and techniques to achieve reproducible results in computational cancer research. We’ll show you the fundamentals of commonly-used approaches in reproducibility that you can apply to increase the impact of your research by making your findings more robust and reliable! To ensure that workshop attendees have a great hands-on experience, there will be a very limited number of seats available.
At the Center for Data-Driven Discovery in Biomedicine (D3b), I lead the Bioinformatics Translational Pediatric Oncology Team, a team of bioinformatics scientists. Our mission is to advance pediatric oncology research and precision medicine through collaboration and development of open-source analytical tools, frameworks, and data resources. In 1998, I lost my four year old cousin John Matthew to a brain tumor we now know was likely a diffuse intrinsic pontine glioma. So, it was bittersweet for me to see the Open Pediatric Brain Tumor Atlas (OpenPBTA) manuscript published in Cell Genomics on the last day of brain tumor awareness month this past year. But let’s rewind.
Writing effective documentation is challenging. Users might not always read every word in the documentation. They might even just scroll past large chunks of text, but we can accommodate those behaviors by structuring and formatting content appropriately.
In 2019, Alex’s Lemonade Stand Foundation (ALSF) established the Single-cell Pediatric Cancer Atlas (ScPCA) through awards for data generation and to create an atlas of single-cell gene expression profiles of pediatric cancers of different types and from different organ sites. The Data Lab launched the ScPCA Portal in 2022 to make uniformly processed, summarized single-cell and single-nuclei RNA-seq data and de-identified metadata available for download. The ScPCA Portal also supports other data modalities, such as bulk RNA-seq, CITE-seq, and spatial transcriptomics. The ScPCA Portal currently hosts data for over 500 pediatric tumor and patient-derived xenograft samples from more than 50 cancer types, and continues to grow. The Data Lab is seeking contributions to the ScPCA Portal from researchers with existing single-cell datasets.
We are excited to announce that our next workshop, Introduction to Single-cell RNA-Seq, will take place in-person from June 13-15, 2023! Data Lab staff will introduce researchers studying pediatric cancer to the R programming language, the Tidyverse R packages for data science, single-cell RNA-seq data analysis, annotating cell types, and more. The 3-day course will take place from 9am-5pm Eastern time in Bala Cynwyd, PA, just outside of Philadelphia. Travel reimbursement (up to a certain amount) is available for qualifying participants.
The Childhood Cancer Data Lab maintains a collection of uniformly processed single-cell data from pediatric cancer clinical samples and xenografts in the Single-cell Pediatric Cancer Atlas (ScPCA) Portal. Although access to preprocessed data saves researchers time, we know that the downloads from the ScPCA Portal are only the starting point. That’s why we’ve created downstream analysis workflows for commonly performed analyses. Instead of writing code wholesale, you can analyze data once you’ve configured these workflows.
We are excited to announce that our next virtual workshop, Introduction to Single-cell RNA-Seq, will run from May 15-19, 2023! In this workshop, Data Lab staff will introduce researchers studying pediatric cancer to the R programming language, the Tidyverse R packages for data science, single-cell RNA-seq data analysis, and annotating cell types.
Last year, the Data Lab launched the Single-cell Pediatric Cancer Atlas (ScPCA) Portal, which today holds uniformly processed single-cell gene expression data obtained from 8 separate labs, over 480 samples, and representing 38 cancer types. The portal is still growing as we continue to receive and process raw data from ScPCA investigators! All uniformly processed data is made available for download on the ScPCA Portal, giving researchers easy access to a growing database of summarized gene expression data and metadata to utilize for their own research. But how exactly did we make sure that all of the data was uniformly processed? And how are we able to ensure uniform processing for incoming samples as the portal continues to grow?
Are you attending the American Association for Cancer Research (AACR) annual meeting in Orlando, FL this year? Visit Alex's Lemonade Stand Foundation (ALSF) at booth 369 in the exhibit hall from April 16-19! You'll find information about ALSF's grants program, the Childhood Cancer Data Lab and more. The Data Lab will also be holding office hours during select time slots.
The Data Lab is excited to announce that our next training workshop will be held virtually from March 13-17, 2023! During this workshop, we will cover advanced topics in the analysis of single-cell RNA-seq data for researchers studying pediatric cancer. The workshop will take place each day from 12-5pm Eastern. Each day consists of lectures and designated time for attendees to work on exercise materials and their own projects with our staff available for consultation. You’ll need a laptop with internet access and to install Zoom and Slack. You will log into an RStudio Server hosted by the Data Lab from your web browser. Pediatric cancer researchers are encouraged to apply now!
In September 2022, the Open Pediatric Brain Tumor Atlas (OpenPBTA) project culminated (for now) in a preprint on bioRxiv. This project, started in late 2019 and co-organized with the Center for Data Driven Discovery in Biomedicine (D3b) at Children’s Hospital of Philadelphia (CHOP), is a collaborative effort to comprehensively describe the Pediatric Brain Tumor Atlas (PBTA), a collection of multiple data types from tens of tumor types (read more about why crowdsourcing expertise for the study of pediatric brain tumors is important here). The project is designed to allow for contributions from experts across multiple institutions. We’ve conducted analysis and drafting of the manuscript openly on the version-control platform GitHub from the project’s inception to facilitate those contributions.
Recently, we told you about the Single-cell Pediatric Cancer Atlas (ScPCA) downstream analysis workflow. This ready-to-go workflow is intended to be used with single-cell and single-nuclei gene expression data available on the ScPCA Portal. We developed this workflow to filter, normalize, and perform dimensionality reduction, as well as incorporate initial clustering results to each processed sample/library object. Now we’re excited to introduce one of our latest offerings for use with ScPCA data, a clustering analysis workflow, which can be applied to datasets after running the filtering, normalization, and dimensionality reduction workflow!
The Data Lab is excited to announce that our next training workshop will be held in-person from January 31-February 2, 2023! During this workshop, we will cover advanced topics in the analysis of single-cell RNA-seq data for researchers studying pediatric cancer. The 3-day course will take place from 9am-5pm Eastern time in Bala Cynwyd, PA, just outside of Philadelphia. Travel reimbursement is available for qualifying participants.
Welcome to the Data Lab’s December Scientific Community Bulletin! Each month we share upcoming opportunities from Alex’s Lemonade Stand Foundation (ALSF), the Data Lab, and other events that we have gathered from a variety of science and research organizations. Subscribe to our blog to be alerted about future Scientific Community Bulletin posts!
In this blog post, I’d like to give an overview of the refine.bio refactoring process and web accessibility considerations. Through this process, our goal is to enhance the site usability and performance by improving the code quality and making the application more accessible. But before going into more details about them, let me provide you a quick history of refine.bio.
Welcome to the Data Lab’s November Scientific Community Bulletin! Each month we share upcoming opportunities from Alex’s Lemonade Stand Foundation (ALSF), the Data Lab, and other events that we have gathered from a variety of science and research organizations. Subscribe to our blog to be alerted about future Scientific Community Bulletin posts!
Here at the Data Lab, we're all about, well, data! We believe that data sharing and accessibility is key to accelerating the research process, and ultimately to improving outcomes for childhood cancer patients. So, we were excited to learn that one of the goals of the NCI/NIH initiative, the Childhood Cancer Data Initiative (CCDI), is to build up a Data Ecosystem that will facilitate pediatric cancer researchers' ability to explore and collect data from disparate resources. Although this Ecosystem is still in the early stages, several components are already being developed and are available for researchers to use! One component that is particularly interesting to us is the CCDI's Childhood Cancer Data Catalog (CCDC).
Welcome to the October Scientific Community Bulletin! Each month we share upcoming opportunities from Alex’s Lemonade Stand Foundation (ALSF), the Data Lab, and other events that we have gathered from a variety of science and research organizations. Subscribe to our blog to be alerted about future Scientific Community Bulletin posts!
Welcome to the September Scientific Community Bulletin! Each month we share upcoming opportunities from Alex’s Lemonade Stand Foundation (ALSF), the Data Lab, and other events that we have gathered from a variety of science and research organizations. Subscribe to our blog to be alerted about future Scientific Community Bulletin posts!
At the Data Lab, we are constantly looking for ways to enhance the tools we build for pediatric cancer researchers. Earlier this year, we launched the Single-cell Pediatric Cancer Atlas portal, a database of uniformly-processed single-cell data from pediatric cancer clinical samples. One way we felt the portal could be even more beneficial to pediatric cancer researchers is with a ready-to-go workflow that takes in single-cell data and prepares it for downstream analyses such as unsupervised clustering.
The Data Lab is excited to announce our next virtual workshop running from September 19-23, 2022! In this workshop, Data Lab staff will introduce researchers studying pediatric cancer to the R programming language, the Tidyverse R packages for data science, single-cell RNA-seq data analysis, and pathway analysis.
The Data Lab teaches data science courses targeted toward pediatric cancer researchers that introduce topics such as analysis of gene expression in bulk and single-cell data and principles of reproducible research. I wrote previously about how we use RStudio Server for our remote courses to simplify setup, and I wanted to write a bit more about some of the instructional practices we use so that our participants get the best experience we can provide. In particular, I wanted to talk about our use of live coding to facilitate active learning, and one of the tools we developed to make our course development just a bit easier.
Welcome to the August Scientific Community Bulletin! Each month we share upcoming opportunities from Alex’s Lemonade Stand Foundation (ALSF), the Data Lab, and other events that we have gathered from a variety of science and research organizations.
Often when building a server-client web application, we will encounter a situation where we want to send requests to our API in the chronological order that they occur on the client. Due to the asynchronous nature of these requests, it might not be possible to send them in the same callback for the event that triggered them. This is because we want to use the response from the previous request to craft our current one. A solution to this problem would be to implement a queue. Instead of calling the API immediately after events occur, implementing a queue ensures the latest data is sent with any request.
Welcome to the July Scientific Community Bulletin! Each month we share upcoming opportunities from Alex’s Lemonade Stand Foundation (ALSF), the Data Lab, and other events that we have gathered from a variety of science and research organizations. Subscribe to our blog to be alerted about future Scientific Community Bulletin posts!
Welcome to the Childhood Cancer Data Lab’s new blog feature, the monthly Scientific Community Bulletin! At the start of each month, we will share upcoming opportunities from Alex’s Lemonade Stand Foundation (ALSF), the Data Lab, and other events that we have gathered from a variety of science and research organizations. Our goal is to promote learning opportunities and highlight some of the excellent resources that our community provides.
At the Data Lab, our science team has a practice where an individual team member shares something that they recently figured out (or didn’t totally figure out yet) on a biweekly basis. We call this short 5-10 minute presentation How I Solved This, and it’s a great way to formally share (often hard-won) knowledge with each other. In this post, we thought we’d share how we solved something with the `renv` package with you.
The Childhood Cancer Data Lab builds resources guided by the most pressing needs of our primary users: pediatric cancer researchers. As the Data Lab's UX Designer, I conduct research activities with scientists like usability evaluations, semi-structured interviews, and card sorts to gain insight into their activities, processes, pain-points, and behaviors. I work with scientists and engineers at the Data Lab to use this information to improve existing products and services or to create new ones.
The Data Lab is excited to announce that our next training workshop is taking place in-person on Friday, June 10, 2022! During this full day workshop, instructors will introduce principles and techniques to achieve reproducible results in computational cancer research. We’ll show you the fundamentals of commonly-used approaches in reproducibility that you can apply to increase the impact of your research by making your findings more robust and reliable!
The Childhood Cancer Data Lab is growing as a resource for pediatric cancer researchers and we have more to offer to our community now, than ever before. Transitioning to our new and improved website is an exciting milestone, and here, we look forward to sharing progress, introducing new initiatives, and cultivating more opportunities to support childhood cancer research. Welcome to our new virtual home!
The Single-cell Pediatric Cancer Atlas (ScPCA) project began in 2019 when Alex’s Lemonade Stand Foundation (ALSF) funded 10 awards for single-cell profiling of pediatric cancer samples. The goal was to produce an atlas of gene expression profiles for a variety of childhood cancer types from different organ sites.
The CCDL team includes science, engineering, and design expertise. Combining these three disciplines in different ways across projects enables us to carry out our mission.
Here at the CCDL we value putting publicly available data to work. For example, we are currently processing and normalizing 1.5 million publicly available gene expression samples totaling ~$1.5 billion research dollars expended.
Like many teams that work with large amounts of external software, we run into issues with our transitive dependencies. In general, transitive dependencies are a hard problem to solve.
Though technology can introduce great benefit into our lives, it is often accompanied by a substantial amount of time and some expected frustration before we can reap the rewards. The time spent learning a new technology is what we usually call a learning curve.
The workshop will last from 9AM to 5PM on October 14th, 15th, and 16th at the CCDL offices at 1429 Walnut St Philadelphia, PA, 19102.
MultiPLIER is a machine learning approach that brings big data to bear on rare diseases. It’s also an example of the scientific approach and ethos of the CCDL, and the publication is a great opportunity to share how the CCDL is developing new technologies to accelerate research into cures for childhood cancers!
The Childhood Cancer Data Lab powered by Alex's Lemonade Stand Foundation is hosting a workshop to introduce childhood cancer researchers to reproducible analysis of bulk and single-cell transcriptomic data.
The Childhood Cancer Data Lab (CCDL), an initiative of Alex's Lemonade Stand Foundation develops tools, trainings, and methods to empower childhood cancer researchers. The work at the CCDL is focused and impactful. There are multiple opportunities and challenges for you to apply and grow your skills as a scientist or as an engineer.
The Childhood Cancer Data Lab powered by Alex's Lemonade Stand Foundation is hosting a workshop to introduce childhood cancer researchers to reproducible analysis of bulk and single-cell transcriptomic data.
At this hands-on, 3-day session held in Houston, researchers learned data science skills that could accelerate their own work. Drawing on skills learned at the workshop, childhood cancer researchers can perform basic analyses of their work to make informed decisions on how to proceed with their own research. Don’t just take our word for it, though. Read more about the workshop’s incredibly valuable benefits through its attendees’ perspectives.
The goal of our refine.bio project is to download, process, and make available gene expression datasets that can be analyzed together, or in parts, depending on a researcher’s need. Childhood cancer researchers need to be able to use data generated through multiple profiling technologies including microarrays and RNA-sequencing.
There are countless log blog posts out there about the benefits of good logging, how to log well, and how much to log. Going through them all can be a real log blog slog. Wouldn't it be cool if you could log like this:logger.info("Something happened!", job=job.id, user=user.id) and get an easily searchable output.
Caffeine is a stimulant that can induce alertness in certain individuals when consumed at an appropriate quantity. Caffeine is often obtained by ingesting caffeine-containing solutions. However, no protocol for obtaining caffeine from dehydrated, roasted beans using materials typically available in a Philadelphia office has been described in the published literature.
Alex’s Lemonade Stand Foundation (ALSF) staunchly believes that stronger scientific sharing practices will accelerate the pace of discovery and finding cures for children with cancer. Robust sharing improves reproducibility, minimizes redundant studies and maximizes our return on research investment.
This year was a big one for the CCDL. In our mission to empower pediatric cancer experts poised for big discoveries with the knowledge, data and methods to reach them we launched a software product, developed and delivered training workshops on single-cell and bulk RNA-seq analysis, and hired our data science team among other milestones.
I’m a scientist at Sage Bionetworks, a nonprofit research organization in Seattle, WA. My work focuses on a family of rare pediatric diseases (NF): neurofibromatosis type 1, type 2, and schwannomatosis.
Our particular process is designed to source opportunities from our team members and external stakeholders, convert those opportunities into a set of potential goals, and then select the goals that we expect will most advance our mission.
The ability to restore scroll position is often critical for website usability. It helps users keep the flow of navigation when going back and forth between different pages. Most modern browsers take care of restoring the scroll position automatically, but it doesn’t always work for Single Page Applications where the content is generated on the client’s side, often asynchronously.
Carnegie Mellon University Libraries is partnering with the Childhood Cancer Data Lab (CCDL), founded by Alex’s Lemonade Stand Foundation, to host a Data Analysis workshop using CCDL materials.
The CCDL will have a team of scientists at the American Association for Cancer Research 2020 Annual Meeting in sunny San Diego! Our team members are excited to talk to researchers studying pediatric cancer at Booth 1601.
To help keep pediatric cancer research moving forward, here are 3 ways the CCDL is helping the research community during this time: refine.bio, virtual workshops, and the Open Pediatric Brain Tumor Atlas project.
We know that pandemic-related university closures mean that the demand for opportunities for pediatric cancer researchers to increase their analytical skills has never been higher. As such, we are delighted to announce a pilot virtual workshop running from May 4-8, 2020!
Here at the Childhood Cancer Data Lab, we value transparency and the practice of open science. Much of the work we’ve done and the products that we build hinge on the generosity and openness of other scientists. In this post, as part of National Brain Tumor Awareness month, we want to talk about a project that our science team has been working on over the last few months (and to do so in a way that aligns with our values).
The workshop will take place on June 22 - 26, 2020 from noon - 5pm Eastern. Each day consists of lectures and designated time for attendees to work on exercise materials and their own projects with CCDL staff available for consultation.
When the CCDL (along with everyone else) realized that we would have to conduct our bioinformatics training workshops remotely, we had to make some quick decisions about how we were going to do it. Most of the instructional materials for our in person workshops were already online, so we knew we had a good base to work from. We just needed to figure how to adapt the live instruction.
At Alex’s Lemonade Stand Foundation’s Childhood Cancer Data Lab, we’re excited to be helping out with an upcoming event hosted by the Children’s Tumor Foundation. If you participate, you may meet members of our team who are mentoring and judging.
The workshop will take place on March 22 - 26, 2021 from noon - 5pm Eastern. Each day consists of lectures and designated time for attendees to work on exercise materials and their own projects with CCDL staff available for consultation
The workshop will take place on June 28- July 2, 2021 from noon to 5pm eastern. Each day consists of lectures and designated time for attendees to work on exercise materials and their own projects with CCDL staff available for consultation.
Hack4Rare is a virtual event that calls for healthcare startups, developers, solutions architects, and hackathon enthusiasts to join researchers, clinicians and patients in developing solutions built around a number of rare diseases including neurofibromatosis, PTEN Hamartoma Tumor Syndrome, RASopathies and Desmoid Tumors.
The workshop will take place on September 20 - 24, 2021 from noon - 5pm Eastern. Each day consists of lectures and designated time for attendees to work on exercise materials and their own projects with CCDL staff available for consultation.
Introducing refine.bio examples. Here, users can access a variety of example analyses implemented in R, such as clustering and heat maps, differential expression analysis, and pathway analysis, for use with refine.bio data.
The workshop will take place on November 1-5, 2021 from noon to 5pm eastern. Each day consists of lectures and designated time for attendees to work on exercise materials and their own projects with our staff available for consultation.
I work at the Childhood Cancer Data Lab, where we use very big data to find cures for childhood cancers. To move data around the internet at very high speeds, we are forced to use a proprietary software suite called Aspera. If somebody could make a Free Software alternative, the future of the internet would be way more awesome! Best of all, you can be the one to do it!
When my daughter Alex was diagnosed with cancer and throughout her battle, we saw how our community of people rallied around our family. No one knew quite how to help, but they were willing to do whatever was needed to ease the burden we faced.
'Work smarter not harder’ is useless advice if you don’t know how to ‘work smarter’. But the Childhood Cancer Data Lab's work and processes may be the smartest I’ve ever had the pleasure of learning and adopting.
The Data Lab will hold our first virtual workshop of the year from March 14-18, 2022!In this workshop, we will introduce researchers studying pediatric cancer to the R programming language, the Tidyverse R packages for data science, single-cell RNA-seq data analysis, and pathway analyses.
At the Data Lab, we are big proponents of automating the boring stuff so we can spend more time thinking about the fun stuff. But how exactly do we do that, and what does it mean to automate the boring stuff?
November marked the final Childhood Cancer Data Lab training workshop for 2021. We held four week-long virtual workshops this year, teaching 88 researchers the data science skills they need to examine their own data.
Before working as a Data Scientist at the Childhood Cancer Data Lab, I spent time in my PhD and post-doctoral fellowship in two very different research environments. Each had their own unique way of doing research. I found that some things worked really well and others were not as successful.

333 E. Lancaster Ave, #414
Wynnewood, PA 19096 USA
© Childhood Cancer Data Lab